



中华医学会神经病学分会帕金森病及运动障碍学组 中国医师协会神经内科医师分会 帕金森病及运动障碍学组
通信作者:刘军、唐北沙、陈生弟、陈海波、王丽娟
【摘要】
多系统萎缩(MSA)是一种成年起病的进展性神经退行性疾病,其病因不明,临床主要表现为自主神经功能障碍、帕金森综合征和小脑综合征等多种组合,早期诊断相对困难,预后不佳。为了更好地规范我国临床医师对MSA的诊断和鉴别,中华医学会神经病学分会帕金森病及运动障碍学组和中国医师协会神经内科医师分会帕金森病及运动障碍学组的相关专家以国内外MSA最新的临床研究结果为依据,结合我国临床实际,对我国MSA诊断标准专家共识进行内容上的更新,以期提高临床医师对MSA诊断的正确率,减少漏诊与误诊。
【关键词】
多系统萎缩;诊断;共识
Expert consensus on diagnostic criteria for multiple system atrophy in China (2022)
Chinese Society of Parkinson′s Disease and Movement Disorders, Parkinson′s Disease and Movement
Disorder Section of Neurologist Branch of Chinese Medical Doctor Association
【Abstract】
Multiple system atrophy (MSA) is an adult‑onset and progressive neurodegenerative disease with unknown etiology and poor prognosis. The clinical symptoms include various combinations of autonomic failure, parkinsonism and cerebellar syndrome. Despite advances in the diagnostic tools for MSA in recent years, early and accurate diagnosis of MSA remains challenging. In order to aid the clinician in recognizing key clinical features and improving diagnostic accuracy of MSA, the Chinese Society of Parkinson′s Disease and Movement Disorders and Parkinson′s Disease and Movement Disorder Section of Neurologist Branch of Chinese Medical Doctor Association updated the expert consensus on the diagnostic criteria for MSA based on China′s practical situations and recently published researches.
【Key words】
Multiple system atrophy; Diagnosis; Consensus
Conflicts of interest: None declared
多系统萎缩(multiple system atrophy,MSA)是一种进展性的神经系统退行性疾病,临床表现为自主神经功能障碍(autonomic failure)、帕金森综合征(parkinsonism)和小脑综合征(cerebellar syndrome)的多种组合[1]。MSA于1969年被首次命名[2],1995年,在美国菲尼克斯召开的共识讨论会议上,根据其主要临床表现被分为黑质纹状体变性、橄榄脑桥小脑萎缩及Shy-Drager综合征3种亚型[3]。由于MSA的自主神经症状多伴有帕金森综合征和(或)小脑综合征的表现,1998年,在美国明尼亚波里召开的共识讨论会议正式确定弃用Shy-Drager综合征,将MSA根据首发运动症状和(或)运动症状的严重程度分为帕金森型多系统萎缩(MSA-parkinsonian type,MSA-P)和小脑型多系统萎缩(MSA-cerebellar type,MSA-C),并根据诊断精确度分为可能的(possible)、很可能的(probable)、确诊的(definite)MSA[4]。2007年,在美国波士顿召开的共识讨论会议延续之前的分类与命名方法,结合技术进展对MSA的诊断标准进行了相应调整[5]。2017年,结合我国实际与新的临床研究依据,我国推出了MSA诊断标准中国专家共识[6]。2022年国际运动障碍协会制订的诊断标准保留了MSA-P和MSA-C的分型,将MSA根据诊断精确度分为神经病理确诊的(neuropathologically established)、临床确诊的(clinically established)、临床很可能的(clinically probable)和前驱可能的(possible prodromal)MSA[1]。
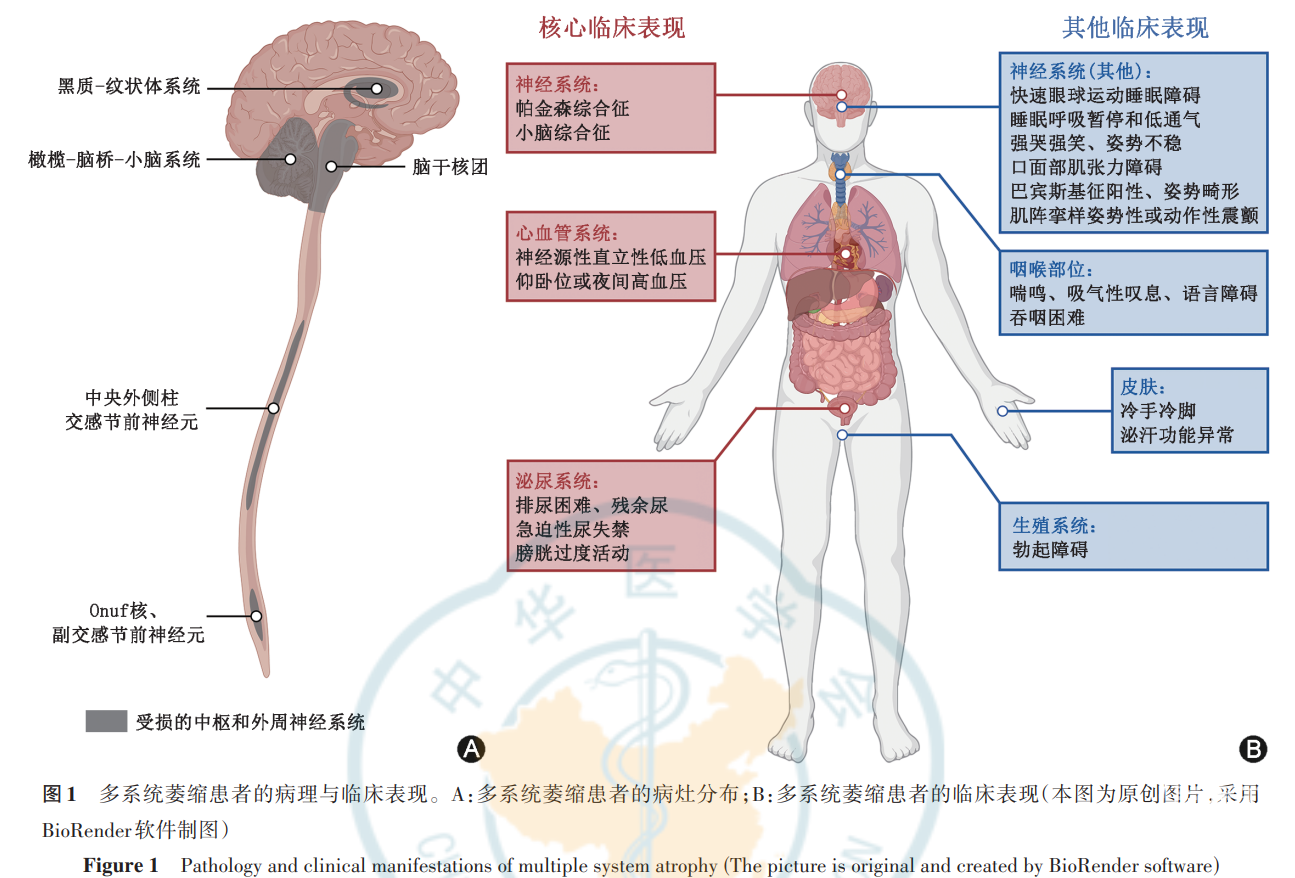
我国尚无明确的MSA流行病学资料。欧美国家的数据显示,MSA的平均患病率为(1.9~4.4)/10万人[7-8],平均发病率为0.6/10万人,50岁以上人群的平均发病率为3.0/10万人[9]。MSA没有明确的危险因素,通常被认为是一种散发疾病,但也有研究发现MSA患者中存在SHC2拷贝数缺失、COQ2突变、SNCA突变[10-12],提示遗传因素可能参与MSA的发病。MSA是一种少突胶质细胞α-突触核蛋白病,其尸检结果通常显示多部位的少突胶质细胞胞质内涵体形成和神经细胞死亡,包括黑质纹状体变性、橄榄脑桥小脑萎缩、脑干多核团神经元丢失、脊髓中央外侧柱、骶髓副交感节前神经元和Onuf核损伤等(图1A)[2,13-14]。
MSA早期诊断困难,预后不佳,临床缺乏准确的生物学标志物。为了更好地规范我国临床医师对MSA的诊断和鉴别诊断,中华医学会神经病学分会帕金森病及运动障碍学组和中国医师协会神经内科医师分会帕金森病及运动障碍学组以国内外新近的临床研究为依据,结合我国实际情况,更新了我国的MSA诊断标准专家共识,以期提高MSA临床诊断的正确率,减少漏诊与误诊。
一、临床表现
MSA临床表现为自主神经功能障碍、帕金森综合征和小脑综合征的多种组合。MSA根据首发运动症状和(或)运动症状严重程度分为MSA-P型和MSA-C型,以帕金森综合征为主的患者为MSA-P型,以小脑综合征为主的患者为MSA-C型[1]。MSA的平均发病年龄为56.2岁[15],46%~61%的患者以运动症状起病,23%~43%的患者以自主神经功能障碍起病,也可同时起病[16-18]。早期出现进展性的严重自主神经功能障碍是MSA的主要特征,并影响患者的生存期[16]。该病进展迅速,约50%的患者在运动症状出现后3年内行走依赖助行器或需要家人扶持,60%的患者5年后需要轮椅,6~8年后患者通常完全卧床[19-21]。欧美国家的数据显示MSA患者起病后的中位生存期是9.8年[15,22],我国最近的研究数据显示MSA患者的中位生存期约为6年[23-24]。MSA常见的死因包括呼吸道感染和猝死[23]。
1. 核心临床表现(图1B):
(1)帕金森综合征:
MSA-P型以帕金森综合征为突出表现,主要表现为运动迟缓,伴肌强直或震颤,但帕金森病(Parkinson’s disease)典型的“搓丸样”震颤少见,多为皮质震颤。MSA患者帕金森综合征进展快,容易出现姿势平衡障碍,往往对多巴胺能药物应答欠佳。帕金森综合征与患者黑质纹状体变性有关。
(2)小脑综合征:
临床表现为步态共济失调、肢体共济失调、小脑性构音障碍和小脑性眼动障碍(持续凝视诱发的水平型或下跳型眼震和扫视性眼动过度),与橄榄脑桥小脑萎缩有关。
(3)泌尿系统功能障碍:
临床表现包括储尿和排尿功能异常,前者表现为尿频、尿急、夜尿、尿失禁,统称为膀胱过度活动征;后者包括排尿费力、尿流间断、尿线细而无力、排尿不尽感、重复排尿等。上述症状与中脑导水管周围灰质腹外侧区和脑桥排尿中枢处的神经元损伤有关(多致储尿功能异常)以及骶髓副交感节前神经元和骶髓前角Onuf运动神经核团的丢失有关(多致排尿功能异常)[13]。在18%的MSA患者中,泌尿系统功能障碍是其唯一的首发表现,在运动症状前平均2.8年出现[25],而在早期帕金森病患者中也常见尿频、尿急、夜尿和排尿困难等症状[26]。尿失禁可见于晚期帕金森病患者,但尿潴留和急迫性尿失禁在早期MSA患者中即可出现。急迫性尿失禁指在没有泌尿系统感染的情况下,MSA患者可突然出现较急的尿意伴不自主漏尿,这两种症状可用于鉴别诊断MSA与帕金森病[27]。
(4)心血管自主神经功能障碍:
临床主要表现为神经源性体位性低血压(neurogenic orthostatic hypotension,nOH),患者出现头晕、晕眩、晕厥,也可出现头颈部疼痛、乏力、恶心、思维减慢、视物模糊、直立性呼吸困难、心绞痛等[28],常伴发夜间或仰卧位高血压[29]。nOH与延髓头端腹外侧兴奋性交感神经元和胸段脊髓中央外侧柱交感节前神经元退行性变有关[13]。
2. 其他非运动症状(图1B):
(1)喘鸣:
患者由于声门裂狭窄在睡眠或清醒时发出高调的吸气声,夜间喘鸣不易被发现。喘鸣的发生机制不明确,有学者推测与声带外展肌瘫痪和声带内收肌肌张力障碍有关,疑核外展运动神经元的丢失和内收运动神经元的保留可能是其潜在的病理解剖基础[30]。喘鸣在MSA患者中的发生率为31.8%,极少见于帕金森病和进行性核上性麻痹(progressive supranuclear palsy,PSP)[31-32]。喘鸣症状对于预测MSA患者的生存期并不明确,但清醒时喘鸣相较于夜间喘鸣提示患者病情更严重[33]。
(2)睡眠障碍:
MSA患者可见多种形式的睡眠障碍[34]。快速眼球运动期睡眠行为障碍(rapid eye movement sleep behavior disorder,RBD)是突触核蛋白病的常见症状,表现为快速眼球运动(rapid eye movement,REM)睡眠期出现梦境演绎,导致受伤或睡眠受扰。RBD的诊断要满足4个条件:
①反复发作睡眠相关发声和(或)复杂动作;
②异常行为经视频多导睡眠监测(video-polysomnogram,vPSG)证实出现于REM睡眠期,或者基于梦境扮演病史推测异常行为出现在REM睡眠期;
③vPSG提示REM睡眠期无肌张力缺失;
④不能以另一种睡眠疾病、精神疾病、药物和物质应用所解释[35]。RBD可能与脑桥背外侧被盖核(sublaterodorsal tegmental nucleus)的退行性变有关[36],可见于41.3%的MSA患者、27.4%的帕金森病患者和7.7%的PSP患者,可用于鉴别MSA与PSP[32,37]。此外,睡眠呼吸暂停和低通气也是MSA患者常见的睡眠呼吸障碍,与脑干中前包钦格复合体(preBötzinger complex)、中缝核、弓状核和疑核的神经细胞丢失有关[38],可能是MSA患者猝死的危险因素[39-40],但目前还不能用于鉴别MSA和其他神经退行性疾病[41]。
(3)吸气性叹息:
临床表现为不自主地深吸气叹息或喘息,常见于夜间非快速眼球运动(non-rapid eye movement)睡眠期的N1和N2睡眠期,与脑干中前包钦格复合体神经元变性有关[42]。吸气性叹息可见于43.6%的MSA-P型患者和3.4%的帕金森病患者[31-32]。
(4)冷手冷脚:
表现为冷手冷脚和肤色变化(紫色或蓝色),按压可发白,恢复较慢,提示血液循环回流不佳。与肢体末端血管交感神经调控障碍有关[43]。
(5)勃起障碍:
临床表现为无法勃起或勃起维持困难导致性功能障碍。与骶髓副交感节前神经元和Onuf运动神经元的损伤有关[44]。勃起障碍是MSA男性患者最常见的自主神经功能异常[17,45],但由于60岁以后正常老年男性勃起功能可迅速减退[46],因此在诊断临床很可能的MSA时,小于60岁患者出现勃起障碍才能作为支持性非运动症状表现。
(6)强哭强笑:
临床表现为患者突然出现不受控制和不合时宜的大哭或大笑,可通过询问病史了解。通常与皮质-脑桥通路受损导致情绪表达障碍有关[47],也有研究表明小脑参与了情绪的调控,尤其在MSA-C型患者当中。强哭强笑可见于23.1%的MSA患者、15.4%的PSP患者,极少见于路易体痴呆(dementia with Lewy bodies,DLB)和帕金森病患者[31-32]。
(7)泌汗功能异常:
MSA患者可表现出节前型无汗症和节前 节后混合型无汗症,MSA患者的泌汗功能障碍较帕 金森病患者更严重[48]。
3. 其他运动症状(图1B):
(1)姿势不稳:
表现为后拉试验时患者退后3步及以上,或在没有检查者帮助的情况下有跌倒的倾向。与黑质纹状体多巴胺能系统和脑桥脚间核(pedunculopontine nucleus)胆碱-谷氨酸能系统退行性变有关[49]。姿势不稳在MSA患者中的发生率为81.6%[15]。姿势不稳通常可见于病程1年内的PSP患者、病程3年内的MSA患者以及病程10年内的帕金森病患者[49-50]。PSP患者病情进展到跌倒的中位潜伏期是6个月,MSA患者是24个月,帕金森病患者是118个月[51]。
(2)口面部肌张力障碍:
表现为不自主的口面部运动异常,可由左旋多巴诱发或加重,不伴或仅伴有轻微的肢体运动障碍。可能因黑质纹状体系统退行性变导致三叉神经、面神经运动障碍[52]。口面部肌张力障碍可见于25.0%的MSA-P型患者和6.8%的帕金森病患者[32]。
(3)咽喉肌运动障碍:
患者可出现构音障碍,表现为发音困难、说话缓慢含糊,通常需要重复对话。患者也可表现为吞咽困难、流涎,需要调整饮食进行适应。与疑核损伤有关。严重的言语障碍可见于45.6%的MSA患者及3.8%的DLB和帕金森病患者[31-32]。
(4)巴宾斯基征阳性:
是上运动神经元损伤的表现,需要排除颅内肿瘤、感染、脑血管疾病、脱髓鞘疾病、代谢性疾病和脊髓型颈椎病等其他原因导致的病理征阳性。
(5)肌阵挛样姿势性或动作性震颤:
当患者维持抵抗重力的姿势或自主运动时,手或手指出现不规律的小幅度震颤,伴刺激敏感的肌阵挛。姿势性或动作性震颤可见于47.4%的MSA-P型患者和5.9%的帕金森病患者[32],肢体肌阵挛更多见于皮质基底节综合征(corticobasal syndrome,CBS)患者,占50%~55%[53-54]。
(6)姿势畸形:
至少包括以下1项,颈部前屈或侧屈、躯干前屈、Pisa综合征和手足挛缩。颈部前屈或侧屈可在一定程度上通过自主或被动运动纠正,Pisa综合征表现为严重的脊柱侧屈,手足挛缩需排除掌腱膜挛缩症(Dupuytren综合征)、CBS或其他原因导致的挛缩。与早中期帕金森病患者相比,颈部前屈、躯干前屈、Pisa综合征和手足挛缩等姿势畸形更常见于MSA患者[31-32,55-56]。
二、辅助检查
辅助检查根据其对临床诊断的价值分为必要性辅助检查和选择性辅助检查。必要性辅助检查对MSA诊断和鉴别诊断以及评估患者的病情有重要意义。选择性辅助检查对诊断MSA有一定的参考价值,临床医生可根据自身设备与人员条件适当完善(表1)。

1. 神经影像学检查:
(1)磁共振成像(magnetic resonance imaging,MRI):
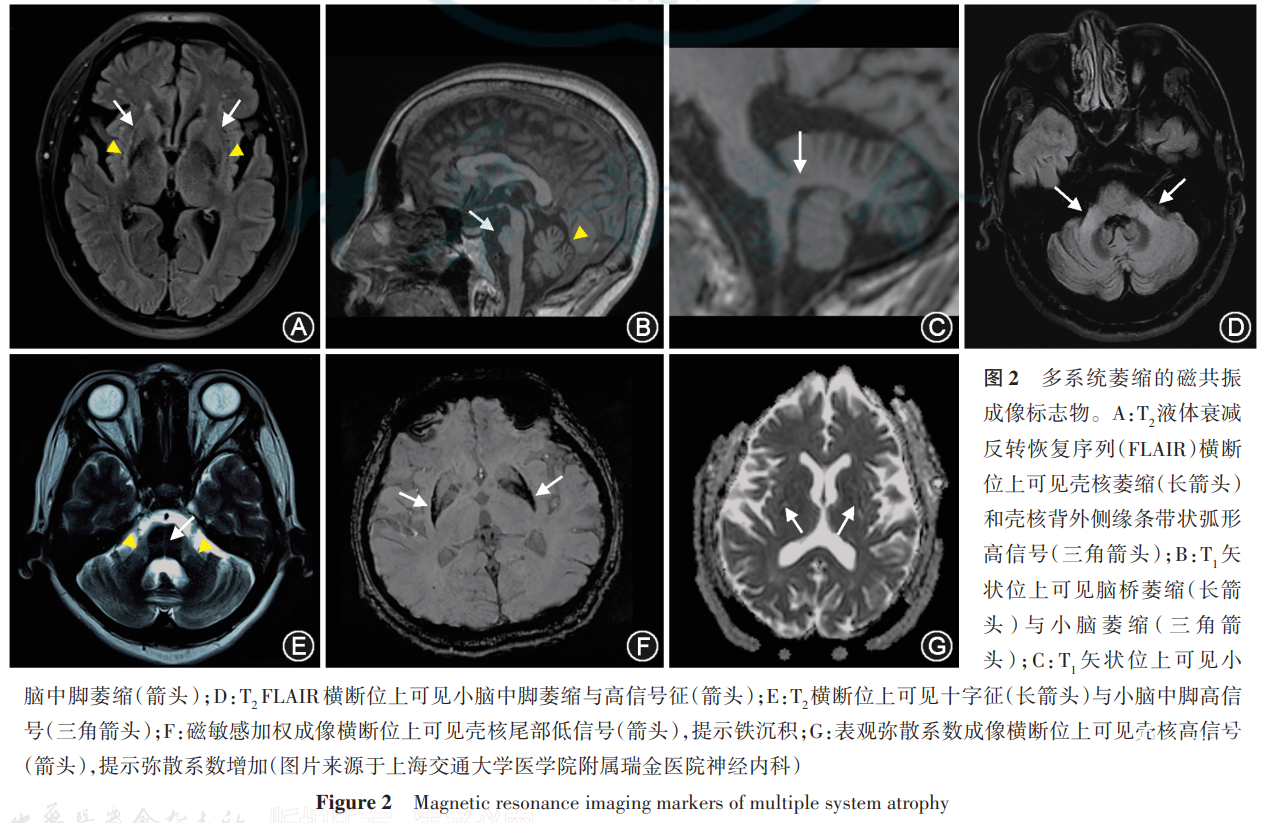
MSA患者表现为常规MRI序列上壳核、脑桥、小脑中脚和小脑萎缩,磁敏感序列上壳核的信号降低,T2序列上脑桥十字形高信号(十字征),弥散加权成像上壳核和小脑中脚弥散系数增加(图2)。MSA-C型患者通常伴有小脑萎缩的表现。由于特发或遗传的小脑萎缩性疾病也可出现小脑萎缩或小脑中脚弥散系数增加,因而小脑萎缩或小脑中脚弥散系数增加不能单独作为临床确诊MSA-C型的影像学依据(表2)[1]。因正常人在3T MRI上也可有T2序列壳核背外侧缘条带状弧形高信号(裂隙征)的类似表现(图2A),且其鉴别MSA-P型与PSP患者的能力不强,不再作为诊断依据,但1.5T MRI上壳核背外侧缘后部的裂隙征仍对MSA-P型和帕金森病的鉴别诊断有一定的参考价值。同时,T2序列上脑桥十字征的分级与MSA-C型中小脑共济失调的严重程度呈正相关[57]。特征性MRI影像学表现可用于鉴别MSA和帕金森病、PSP、散发性成年起病型共济失调(sporadic adult-onset ataxia,SAOA),但在疾病早期敏感度不足[58]。基于体素的形态学测量、磁化传递率、神经黑色素成像、多模态MRI等磁共振技术也被尝试用于MSA的鉴别诊断[58]。


(2)放射示踪成像:
18氟-氟代脱氧葡萄糖-正电子发射体层摄影(18F-fluorodeoxyglucose-positron emission tomography,18F-FDG-PET)显示MSA患者壳核(后侧)、脑桥和小脑处于低代谢,可用于鉴别MSA与帕金森病[58]。多巴胺转运体-单光子发射体层摄影(dopamine transporter-single photon emission computed tomography,DAT-SPECT)可进行突触前多巴胺能成像,MSA-C型患者DAT摄取能力下降,SAOA患者DAT摄取能力保持正常,可用于早期鉴别诊断MSA-C型和SAOA[59],但在鉴别诊断MSA-P型和帕金森病时证据不足[60-61]。而多巴胺转运体-正电子发射体层摄影(dopamine transporter-positron emission tomography,DAT-PET)由于具备较高的分辨率,可将纹状体进行区域划分:腹侧纹状体、尾状核前部、尾状核后部、壳核前部、壳核后部和壳核腹侧部。研究发现与正常对照相比,帕金森病和MSA-P型患者在上述各个区域DAT结合率明显下降。与帕金森病患者相比,MSA-P型患者壳核腹侧部的DAT结合率下降更明显且出现更早,而壳核后部/壳核腹侧部的DAT结合率比值则更高,该比值用于鉴别诊断的敏感度较高,但特异度较低[62],也有研究发现深度神经网络可利用DAT-PET数据对MSA与帕金森病进行鉴别[63],因此,DAT-PET用于鉴别诊断不同帕金森综合征的效果还有待进一步研究。纹状体部位突触后膜D2多巴胺受体PET显像显示MSA-P型患者的D2受体水平显著下降,帕金森病患者D2受体正常或代偿性升高,可用于鉴别诊断MSA-P型和帕金森病[64-65]。
(3)经颅超声成像:
有研究报道MSA-P型患者黑质回声多正常,黑质高回声能在一定程度上鉴别MSA-P型与帕金森病,但各项研究结果结论不一致,其鉴别诊断的敏感度与特异度欠佳[66-67]。
2. 自主神经功能检查:
(1)泌尿系统功能评价:
①残余尿:膀胱超声、尿流动力学试验可用于测量残余尿,残余尿量超过100ml是诊断临床确诊的MSA的重要指标[27]。随着病情进展,显著的残余尿增多可用于鉴别MSA与帕金森病、SAOA,但不能鉴别MSA与PSP[27,68]。
②尿流动力学试验:主要监测尿流率、膀胱收缩指数、膀胱顺应性等指标。尿流率根据测量方法分为自由尿流率和压力尿流率,MSA患者的最大尿流率(maximum flow rate,Qmax)通常降低,表现为自由尿流率的Qmax<4.5ml/s,压力尿流率的Qmax<3.5ml/s。膀胱收缩指数(bladder contractility index,BCI)可准确反映膀胱的收缩功能(BCI=最大尿流率时的逼尿肌压力+5×压力尿流率试验下的最大尿流率),BCI<36.5提示MSA,可用于鉴别MSA与帕金森病。膀胱顺应性可利用膀胱容量变化与逼尿肌压力变化的比值进行计算,比值<20ml/mmH2O(1mmH2O=0.0098kPa)称为膀胱顺应性下降,常见于MSA患者[69]。
③逼尿肌括约肌协同障碍:通过尿流动力学试验联合肌电图或膀胱尿道造影诊断,表现为排尿过程中逼尿肌的收缩伴随尿道外括约肌电活动升高,或影像上出现尿道狭窄,提示MSA。帕金森病患者不常出现逼尿肌括约肌协同障碍[41,70],但常出现逼尿肌过度活动,表现为膀胱充盈过程中逼尿肌压力的不自主升高,伴随少量漏尿[69]。
(2)心血管自主神经功能评价:
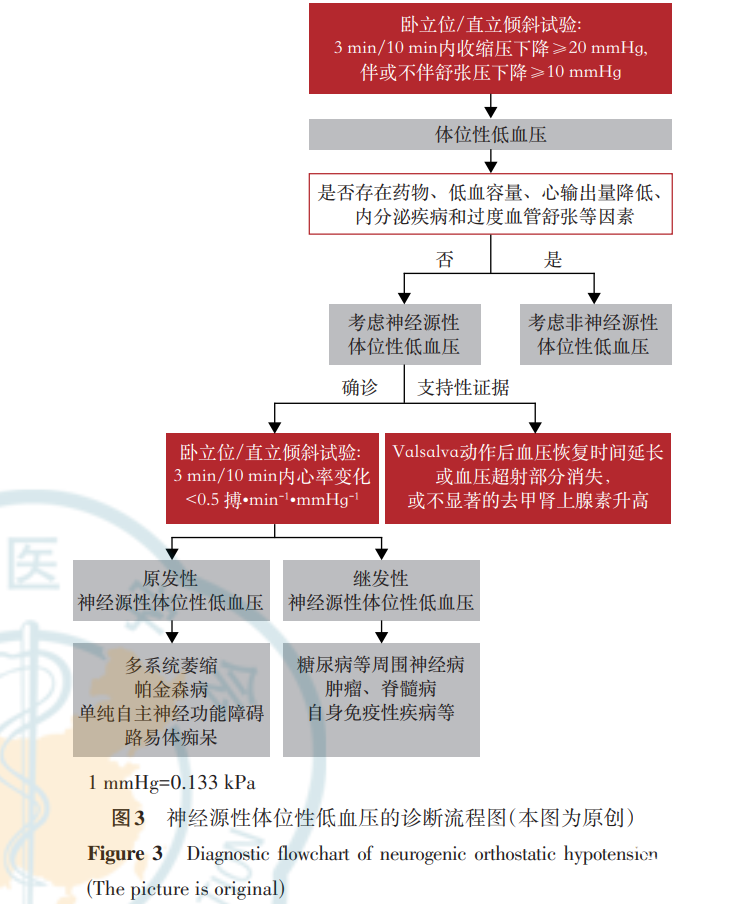
卧立位试验或直立倾斜试验可用于评估体位性低血压,直立倾斜试验的倾斜角度规定为60°~80°[71]。诊断体位性低血压时要求患者在卧立位试验/直立倾斜试验中直立3min或10min内(直立不同时间的诊断级别不同,具体见表2,3[1])收缩压下降≥20mmHg(1mmHg=0.133 kPa),伴或不伴舒张压下降≥10mmHg,舒张压变化不作为必要条件。由于舒张压下降≥10mmHg诊断体位性低血压特异度较差,因而仅舒张压下降≥10mmHg不能作为体位性低血压的诊断依据。试验期间需排除药物(降压药、抗抑郁药)、低血容量(脱水、急性失血)、心输出量降低(缩窄型心包炎、心肌病、主动脉狭窄)、内分泌疾病(肾上腺功能不全、无功能肾上腺嗜铬细胞瘤)和过度血管舒张(系统性肥大细胞增多症、类癌综合征)等非神经源性因素导致的体位性低血压。鉴别患者是否为nOH,首选卧立位/直立倾斜试验观察患者3min或10min内的心率变化,若心率变化<0.5搏·min-1·mmHg-1可确诊为nOH,评估期间避免使用影响心率的药物(如β受体阻滞剂等)。Valsalva动作后血压恢复时间延长或血压超射部分消失以及不显著的去甲肾上腺素(norepinephrine,NE)水平升高等可进一步支持nOH的诊断(图3)[72]。此诊断标准与2008年Gilman等[5]提出的诊断标准(血压下降≥30/15mmHg)相比,在诊断特异度上相似,敏感度有显著提高。此外,24h动态血压监测可用于评估MSA患者的夜间高血压。
(3)vPSG:
vPSG在RBD的诊断中发挥重要作用,RBD经vPSG证实其异常行为出现于REM睡眠期,且REM睡眠中无肌张力缺失[35]。研究报道vPSG监测中的下颌肌电图强直电位有助于鉴别伴有RBD的MSA-P型患者与帕金森病患者[73]。睡眠呼吸暂停要求口鼻温度传感器或气道正压(positive airway pressure)设备气流或替代呼吸暂停传感器信号曲线峰值较事件前基线值下降≥90%,持续时间≥10s,睡眠低通气要求鼻压力传感器、气道正压设备气流或替代低通气传感器信号曲线峰值较事件前基线值下降≥30%,持续时间≥10s,且氧饱和度相较于事件前基线下降≥3%,或该事件导致患者觉醒[74]。吸气性叹息在vPSG上表现为鼻压力传感器、口鼻温度传感器和胸腹呼吸感应体积描记(respiratory inductance plethysmography)绑带的信号曲线峰值较事件前基线升高至少2倍,同时在鼾声通道上可见记录,在视频中可闻及吸气音。为避免呼吸暂停后恢复性深吸气的干扰,呼吸暂停后的吸气性叹息不纳入考虑[42]。
(4)喉镜检查:
喘鸣可通过日间临床表现或夜间vPSG记录下的高调吸气声来诊断,纤维喉镜可发现患者声带出现外展障碍或矛盾内收运动,并排除器质性声带病变(如肿块或瘢痕)或非MSA神经源性声带功能障碍。若患者日间喉镜检查正常但临床怀疑喘鸣,则进行药物诱导下睡眠内窥镜检查[33]。
(5)123碘-间碘苄胍-心肌显像(123I-MIBG-scintigraphy):
MIBG和NE递质有相似的摄取、储存和释放机制,因而123I-MIBG心肌摄取检查反映节后交感神经突触前末梢功能。心肌123I-MIBG的摄取有2种方式,1种为高浓度时的弥散,发生在早期(注射15~30min后);另一种为低浓度时的延迟摄取(注射3~4h后),反映节后交感神经突触前末梢的摄取能力。MSA患者延迟期心肌/膈肌显像剂摄取比值通常正常,而帕金森病患者的摄取比值下降,提示心肌节后交感纤维功能障碍[75]。影响NE转运和储存的药物、心脏结构性病变、糖尿病导致的末端神经病变也可能导致心肌摄取显像剂减少。
(6)盆底神经生理学:
在排除了可能导致慢性神经再生的马尾损伤、盆底手术和产科盆底撕裂的情况下,肛门外括约肌肌电图上若有超过50%的运动单元电位(motor unit potential)单个持续时间>10ms,或平均持续时间>10ms,则提示患者为MSA-P型[76],但该表现也可见于PSP和晚期帕金森病患者。6年以上病程的患者若表现出正常的肛门外括约肌肌电图,通常不考虑诊断为MSA。
(7)发汗试验:
体温调节汗液试验(thermoregulatory sweat testing,TST)可评估患者整体的出汗功能,定量催汗轴突反射试验(quantitative sudomotor axon reflex testing,QSART)可评估节后交感排汗纤维的功能[48]。TST和QSART的组合可用于判断节前、节后或混合型泌汗功能异常。
3. 左旋多巴疗效评定:
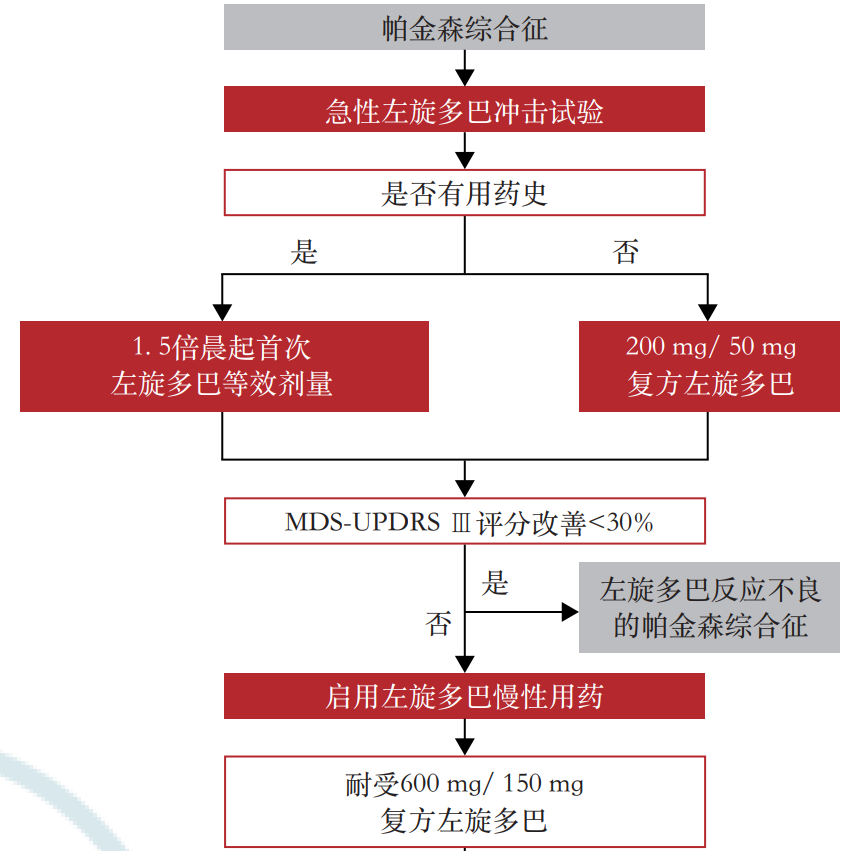
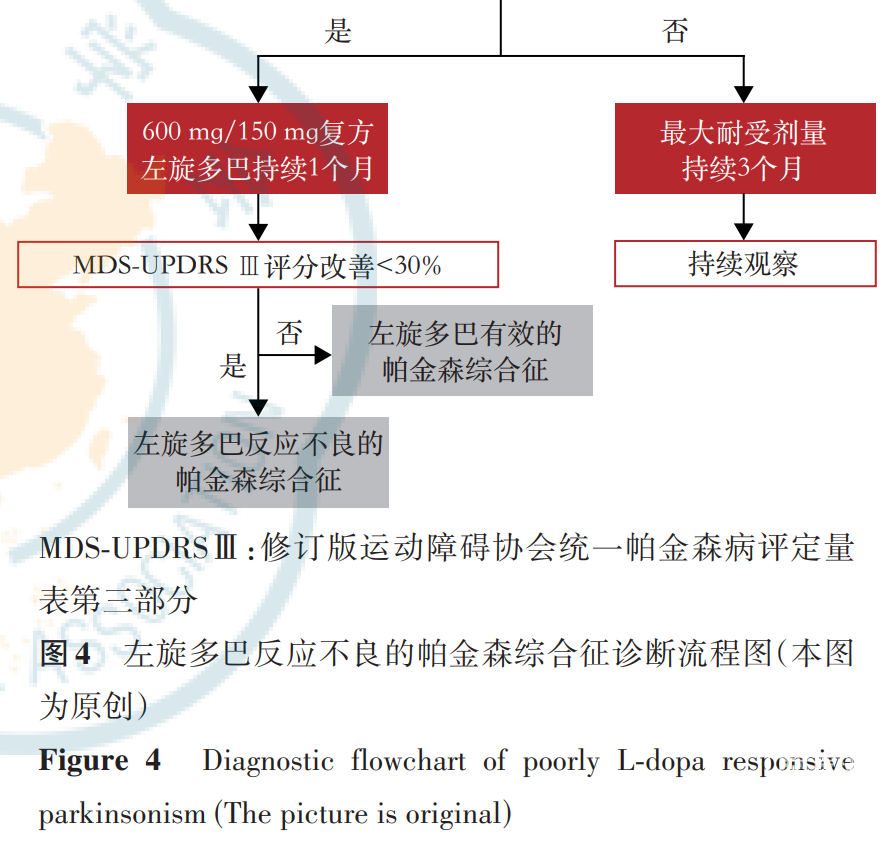
MSA-P型患者可通过判断其对左旋多巴的疗效来辅助诊断,可通过服药史获得相关信息,对于无相关病史的患者或服药效果不明确的患者(图4),可进行急性左旋多巴冲击试验[77]。具体流程如下:要求停用左旋多巴12h,停用单胺氧化酶B抑制剂24h,停用多巴胺受体激动剂72h,并在试验开始前2d口服多潘立酮(20mg,3次/d)以减轻左旋多巴诱导的呕吐和体位性低血压。对于初次就诊未服药的患者,使用200mg/50mg的复方左旋多巴试剂(左旋多巴/外周多巴脱羧酶抑制剂),对于有服药史的患者,使用晨左旋多巴等效剂量的1.5倍,若该剂量仍小于200mg/50mg的复方左旋多巴试剂,则直接服用200mg/50mg的复方左旋多巴试剂,空腹服药。在服药前和服药后30min、1.0h、1.5h和2.0h(或患者自身感觉运动功能恢复最佳的时候)分别评定修订版运动障碍协会统一帕金森病评定量表Ⅲ(Movement Disorder Society-sponsored revision of the Unified Parkinson’s Disease Rating Scale Part Ⅲ,MDS-UPDRSⅢ)评分,若评分改善<30%,提示为左旋多巴反应不良的帕金森综合征。但由于63%的MSA患者运动症状在早期可因服用左旋多巴产生>30%的评分改善[78],因此评分改善>30%的患者需要继续观察其对左旋多巴长期用药的反应。对于耐受每日600mg/150mg复方左旋多巴试剂的患者,持续用药1个月,比较患者1个月后晨起服药与1个月前药物洗脱状态下的MDS-UPDRSⅢ评分,若评分改善<30%,提示为左旋多巴反应不良的帕金森综合征,对于不耐受的患者则较难判断左旋多巴药物对患者的疗效,可持续观察患者对每日最大耐受剂量(持续3个月)药物的反应[1]。
4. 生化检查:
(1)仰卧位血浆NE水平:
测量前需停用咖啡因、酒精、尼古丁12h,停用对乙酰氨基酚5d,测量血浆左旋多巴含量<15nmol/L,患者仰卧平躺15min后,于清晨空腹抽血[79-81]。MSA患者通常表现出正常的血浆NE水平(100pg/ml),而伴nOH的帕金森病/路易体病患者或因自身免疫性疾病导致自主神经功能障碍的患者,血浆NE水平通常降低。在纯自主神经功能障碍(pure autonomic failure,PAF)的患者中,仰卧位血浆NE水平>100pg/ml在预测PAF转化为MSA上有较高的敏感度和特异度[82]。
(2)蛋白质错误折叠循环扩增(protein misfolding cyclic amplification,PMCA)/实时震动诱导转换(real-time quaking-induced conversion,RT-QuIC):
利用PMCA技术扩增脑脊液中的α-突触核蛋白寡聚体后,通过硫黄素T荧光来反映扩增后的纤维含量。MSA患者脑脊液中的α-突触核蛋白扩增后硫黄素T荧光值低于2000,而帕金森病和DLB患者的硫黄素T荧光值明显升高,可用于鉴别MSA与帕金森病、DLB[83]。通过RT-QuIC技术扩增脑脊液中的α-突触核蛋白寡聚体后,MSA患者脑脊液的硫黄素T荧光同样显著低于帕金森病和DLB患者[84]。若PAF患者脑脊液α-突触核蛋白经PMCA扩增后的硫黄素T荧光保持在150~2000AU,提示PAF会向MSA转变[85]。
(3)脑脊液中的神经纤维轻链(neurofilament light chain,NfL):
酶联免疫吸附试验检测脑脊液中的NfL水平>1400pg/ml提示患者为MSA,帕金森病和DLB患者的NfL水平较低[83],若PAF患者脑脊液NfL水平>1400pg/ml,则提示PAF会向MSA转变[85]。
(4)皮肤活组织检查(活检)免疫组织化学试验:
MSA患者中α-突触核蛋白通常沉积于体神经纤维末端,帕金森病患者的α-突触核蛋白通常沉积于自主神经纤维末端,因此对皮肤小动脉、汗腺、立毛肌等自主神经调控的组织进行活检可发现,MSA患者无明显的α-突触核蛋白沉积,帕金森病患者则有显著的α-突触核蛋白沉积[86-87]。
5. 基因检测:
MSA无明确致病基因。为鉴别遗传性共济失调与MSA-C型患者,可筛查共济蛋白基因(frataxin,FXN)、突触核膜蛋白基因(synaptic nuclear envelope protein 1,SYNE 1)、Aprataxin基因(APTX)、Senataxin基因(SETX)、ATXN1、ATXN2、ATXN3、ATXN7、钙离子电压门控通道亚单位α1A基因(calcium voltage-gated channel subunit alpha1 A,CACNA1A)、TATA结合蛋白基因(TBP)、脆性X信使核糖核蛋白1基因(fragile X messenger ribonucleoprotein 1,FMR1)等[88]。
三、评估量表
欧洲多系统萎缩研究组于2004年建立了统一多系统萎缩评价量表(Unified Multiple System Atrophy Rating Scale,UMSARS),UMSARS包括病史回顾、运动检查、自主神经功能检查和整体失能程度4个项目,评分越高提示症状越重,是评估MSA病情进展首选的半定量评估量表[89]。但随着UMSARS的广泛应用,其局限性也逐渐显露:
(1)UMSARS中部分项目能较好地反映疾病进展,尤其是与运动功能评估相关的项目,而与自主神经功能评估相关的项目对疾病进展不敏感。
(2)UMSARS评估在疾病晚期存在天花板效应,不能有效反映晚期疾病进展。
(3)UMSARS对部分MSA相关的症状缺乏评估,如喘鸣、睡眠障碍、流涎、发声障碍、挛缩疼痛等。
(4)UMSARSⅢ要求记录站立2min后的血压和心率,但MSA诊断标准评估的是站立/直立倾斜试验3min内的变化[90-91]。这提示我们MSA患者的评估还有赖更加全面和准确的评估量表。
其他运动功能评定量表:MDS-UPDRS 于2008年由UPDRS扩增修改而来,分为4个部分,包括日常生活中的非运动表现、运动表现、运动评估和运动并发症,MSA患者常用MDS-UPDRS来评估运动改善情况。MSA患者还可利用Tinetti步态和平衡测试(Tinetti Gait and Balance Test)、Berg平衡量表(Berg Balance Scale)、步态及摔倒问卷(Gait and Falls Questionnaire)、改良Webster量表(Modified Webster Scale)来评估运动功能。
非运动功能评定量表:非运动症状筛查量表(Non-Motor Symptoms Questionnaire)、帕金森病结局量表-自主神经系统功能障碍(Scales for Outcomes in Parkinson's Disease-Autonomic Dysfunction)、嗅棒试验16(Sniffin' Sticks Test-16)、Wexner便秘评分(Wexner Constipation Score)、快速眼球运动期睡眠行为障碍筛查量表(Rapid Eye Movement Sleep Behavior Disorder Screening Questionnaire)、匹兹堡睡眠质量指数(Pittsburgh Sleep Quality Index)、帕金森病睡眠质量量表 (Parkinson's Disease Sleep Scale)、39项帕金森病调查问卷(Parkinson's Disease Questionnaire-39)可用于评估患者的非运动功能和生活质量,利用汉密尔顿抑郁量表(Hamilton Depression Rating Scale)、汉密尔顿焦虑量表(Hamilton Anxiety Rating Scale)、蒙特利尔认知评估(Montreal Cognitive Assessment)、简易精神状态检查量表(Mini-Mental State Examination)、额叶功能评分(Frontal Assessment Battery)等评估患者的精神心理状态。
此外,利用嗓音障碍指数(Voice Handicap Index)、吞咽筛查量表(Eating Assessment Tool)、标准吞咽功能评价量表(Standardized Swallowing Assessment)评估患者的咽喉功能。
四、诊断
目前MSA的诊断主要参考2022年MDS提出的诊断标准[1]和2017版多系统萎缩诊断标准中国专家共识[6],结合我国MSA的相关临床研究,将MSA根据诊断精确度分为神经病理确诊的、临床确诊的、临床很可能的和前驱可能的MSA。
1. 神经病理确诊的MSA:
相当于之前诊断标准中确诊的(definite)MSA,尸检病理结果显示中枢神经系统大量胶质细胞胞质内含有α-突触核蛋白阳性的包涵体(glial cytoplasmic inclusions),并存在纹状体黑质或橄榄桥脑小脑结构的神经退行性改变。
2. 临床确诊的MSA:
需要满足散发、进展性、成年起病(>30岁)的基本特征,同时具有核心临床表现,至少存在2项支持性临床表现,至少存在1项MRI标志,不存在排除性的临床表现(表2)[1]。
3. 临床很可能的MSA:
需要满足散发、进展性、成年起病(>30岁)的基本特征,同时具有核心临床表现,至少存在1项支持性临床表现,不要求MRI标志,不存在排除性的临床表现(表2)[1]。
4. 前驱可能的MSA:
需要满足散发、进展性、成年起病(>30岁)的基本特征,必须具有至少1项临床非运动特征(准入标准),同时具有至少1项临床运动特征,不存在排除性临床表现(表3)[1]。

五、鉴别诊断
1. 帕金森病:
帕金森病患者的帕金森综合征对多巴胺能药物的疗效明确且显著有效,常伴静止性震颤与嗅觉减退。MSA-P型患者对左旋多巴疗效欠佳,可伴有姿势性与动作性震颤,嗅觉减退少见,可早期出现进展性的严重自主神经功能障碍。借助MRI、18F-FDG-PET、D2受体成像、经颅超声成像、123I-MIBG-心肌显像进行鉴别。
2. PSP:
PSP患者通常表现出垂直核上性凝视麻痹或垂直扫视变慢,较MSA更早出现反复的自发性摔倒与冻结步态,可伴有左旋多巴反应不良的帕金森综合征和认知障碍,通常无nOH。MSA-P型患者可表现出小脑性眼球运动障碍,姿势平衡障碍较PSP进展稍慢,认知障碍少见,而伴有显著的自主神经功能障碍显著,可出现RBD。两者可借助神经影像学检查进行鉴别。
3. DLB:
痴呆是DLB患者的必要特征,可表现为早期注意力、执行功能和视觉空间能力的损害,在注意力与警觉方面呈波动性认知障碍,有反复发作的视幻觉,可伴有左旋多巴反应不良性帕金森综合征和RBD。MSA-P型患者多不伴有痴呆、波动性认知障碍与视幻觉,可早期出现进展性的严重自主神经功能障碍。两者可借助结构MRI和分子显像进行鉴别。
4. CBS:
CBS患者常出现不对称的四肢肌强直或运动不能、肌张力障碍和肌阵挛,同时可出现口颊或肢体失用、皮质复合感觉丧失和异己肢体。MSA-P型患者四肢肌强直多对称,且少见口颊或肢体失用、皮质复合感觉丧失以及异己肢体等,两者可借助结构MRI和分子显像进行鉴别。
5. SAOA:
SAOA主要指成年发病的、没有明确家族史的进展性共济失调患者,现病因未明,患者疾病进展相对较慢,不出现严重的自主神经功能障碍。MSA-C型患者可早期出现进展性的自主神经功能障碍,借助MRI、DAT-SPECT进行鉴别。
6. 脆性X相关震颤/共济失调综合征(fragile X-associated tremor/ataxia syndrome,FXTAS):
FXTAS多在50岁以后发病,为X连锁遗传,男性多见,以意向性震颤、小脑性共济失调步态、帕金森综合征候群、认知功能减退、周围神经病及自主神经功能障碍为主要临床表现。头颅MRI可见小脑中脚白质病变。基因检测可发现FMR1基因突变。MSA-C型患者认知障碍少见。
7. 脊髓小脑性共济失调(spinocerebellar ataxia,SCA):
SCA是一组由基因突变导致的小脑、脑干、脊髓退行性变,以进行性小脑性共济失调、构音障碍为主要临床表现,可伴有锥体外系症状、锥体束症状、认知障碍、视力障碍、肌阵挛、眼肌麻痹等其他神经系统体征的遗传性疾病。患者可有家族史,不出现严重的自主神经功能障碍。影像上SCA患者与MSA-C患者有重叠,通过基因筛查明确诊断。
六、总结
本共识较上版共识主要在以下内容方面进行了更新:第一,更加详细地阐述了MSA的临床表现以及相应的临床特点在MSA与帕金森病和其他帕金森综合征中的鉴别意义;第二,新增了MSA多系统受损的解剖部位以及对应的MSA的临床表现,并增加图片以更加直观地展现MSA的临床特征和影像学特征;第三,根据2022年MDS提出的诊断标准[1],将MSA根据诊断精确度分为神经病理确诊的、临床确诊的、临床很可能的和前驱可能的MSA;第四,增加了详细的MSA自主神经评估方法,并制订了左旋多巴有效性评估的流程和nOH的诊断流程等,以提高临床工作中诊断MSA的可操作性;第五,增加了可能提升MSA诊断敏感度和特异度的生化检查方法。
MSA是成年起病的进展性神经退行性疾病,病因不明,预后不佳。临床表现为自主神经功能障碍、帕金森综合征和小脑综合征的多种组合。临床上结合影像学、检验等技术可提高检查的敏感度与特异度。发病早期需与帕金森病、DLB、PSP、SAOA进行鉴别诊断。目前以对症治疗为主。目前的研究也在尝试多种方法提高MSA的诊断准确性以及延缓MSA的疾病进展。
执笔 谭玉燕(上海交通大学医学院附属瑞金医院神经内科)、黄思佳(上海交通大学医学院附属瑞金医院神经内科)
专家组成员(按姓氏笔画排序)万新华(中国医学科学院北京协和医院)、王丽娟(广东省人民医院)、王坚(复旦大学附属华山医院)、王含(中国医学科学院北京协和医院)、王青(南方医科大学珠江医院)、王春喻(中南大学湘雅二院)、王振福(解放军总医院第二医学中心)、王晓平(上海交通大学附属同仁医院)、王涛(华中科技大学同济医学院附属协和医院)、王铭维(河北医科大学第一医院)、卢晓东(杭州师范大学附属医院)、叶民(南京医科大学附属明基医院)、叶钦勇(福建医科大学附属协和医院)、乐卫东(四川省人民医院)、冯涛(首都医科大学附属天坛医院)、田玉玲(山西医科大学第一医院)、刘卫国(南京脑科医院)、刘艺鸣(山东大学齐鲁医院)、刘春风(苏州大学附属第二医院)、刘军(上海交通大学医学院附属瑞金医院)、刘振国(上海交通大学医学院附属新华医院)、卢宏(郑州大学第一附属医院)、朱晓东(天津医科大学总医院)、杨新玲(新疆医科大学第二附属医院)、肖勤(上海交通大学医学院附属瑞金医院)、邹海强(解放军南部战区广州总医院)、吴云成(上海交通大学医学院附属第一人民医院)、吴逸雯(上海交通大学医学院附属瑞金医院)、张玉虎(广东省人民医院)、张克忠(南京医科大学第一附属医院)、张宝荣(浙江大学医学院附属第二医院)、张振涛(武汉大学人民医院)、陈生弟(上海交通大学医学院附属瑞金医院)、陈先文(安徽医科大学附属第一医院)、陈伟(上海交通大学医学院附属第九人民医院)、陈玲(中山大学附属第一医院)、陈彪(首都医科大学宣武医院)、陈海波(北京医院)、陈蕾(天津市环湖医院)、邵明(广州市惠爱医院)、沈岳飞(广西医科大学第一附属医院)、苏闻(北京医院)、罗晓光(深圳市人民医院)、罗蔚锋(苏州大学附属第二医院)、罗巍(浙江大学医学院附属第二医院)、承欧梅(重庆医科大学附属第一医院)、金莉蓉(复旦大学附属中山医院)、胡兴越(浙江大学医学院附属邵逸夫医院)、胡盼盼(安徽医科大学第一附属医院)、徐评议(广州医科大学附属第一医院)、唐北沙(中南大学湘雅医院)、顾平(河北医科大学第一医院)、高中宝(解放军总医院)、常颖(吉林大学中日联谊医院)、郭纪锋(中南大学湘雅医院)、陶恩祥(中山大学孙逸仙纪念医院)、黄卫(南昌大学附属第二医院)、商慧芳(四川大学华西医院)、梁战华(大连医科大学附属第一医院)、崔桂云(徐州医科大学附属医院)、焦玲(贵州医科大学附属医院)、程焱(天津医科大学总医院)、谢安木(青岛大学附属医院)、靳令经(同济大学附属养志康复医院)、蔡晓杰(北京医院)、熊念(华中科技大学同济医学院附属协和医院)、薛峥(华中科技大学同济医学院附属同济医院)
利益冲突 所有作者声明无利益冲突
声明:脑医汇旗下神外资讯、神介资讯、神内资讯、脑医咨询、Ai Brain 所发表内容之知识产权为脑医汇及主办方、原作者等相关权利人所有。
投稿邮箱:NAOYIHUI@163.com
未经许可,禁止进行转载、摘编、复制、裁切、录制等。经许可授权使用,亦须注明来源。欢迎转发、分享。
