



孤独症谱系障碍(Autism spectrum disorder, ASD)是一类以社会交往障碍、重复刻板行为和高度狭隘兴趣为基本特征的神经发育障碍性疾病,可伴有癫痫、智力落后、睡眠障碍、胃肠道问题等多种共患病。尽管ASD的病因和临床症状具有高度异质性,近年来对ASD风险基因和交叉神经科学方面的进展,有望为快速识别ASD关键的生物学特征和寻找潜在的治疗靶点奠定基础。
来自加州大学旧金山分校Weill神经科学研究所的Matthew W. State教授课题组较为系统全面地综述了关于ASD基因遗传和交叉神经科学方面的进展,相关综述发表于2022年04月19日发表在Nature Reviews Neuroscience杂志(IF: 34.870)上,题为“Genomics, convergent neuroscience and progress in understanding autism spectrum disorder”。


Matthew W. State教授
Matthew W. State教授就职于加州大学旧金山分校Weill神经科学研究所精神病学和行为科学系,其实验室致力于识别ASD风险基因,并利用系统生物学方法来表征这些基因在人类大脑发育过程中的空间和时间交叉性。
近年来相关研究工作发表于Neuron(2021)、Cell Reports(2018)等杂志上。
ASD风险基因和位点的发现
全外显子测序(WES)
过去二十年里,在鉴定特发型ASD的风险基因和位点方面取得巨大进展。针对罕见变异的微阵列和WES研究已经鉴定了大约十几个拷贝数变异 (CNV)位点和100多个基因,并且这个数字还在不断增加。这种方法旨在识别非亲缘关系的患者中出现同一基因变异,尤其是新发突变(de novo)。总的来说,从WES中鉴定出的罕见变异对生物学研究和临床患者评估均具有重要意义。目前,WES对临床患者评估的贡献度在10% ~ 50%之间。
全基因组测序(WGS)
相比于WES,WGS在识别ASD风险基因和位点方面的贡献相对有限。WGS在检测方法上类似于WES,但其检测的范围更加广泛,其包含了基因的非编码区域。但是,目前由于以下几个因素限制了WGS的应用:1. WGS数据的分析和储存对硬件设备的要求高;2. 目前对非编码区变异可能带来的风险解释尚未清楚。未来需要更大规模的队列研究来明确个体非编码变异的风险。
共同变异相关的发现
目前,大多数ASD风险基因都来自对具有较大效应的罕见变异,但许多强有力的研究表明,小效应的常见变异具有大多数人群风险。最近,一项纳入18,000多个病例和近28,000名对照的研究成功确定了5 个具有中等效应的风险基因。
ASD相关基因的数量
目前,基于普遍接受的外显子组(exome-wide)显著P值的阈值界定, ASD的风险基因有26个,进一步调整最高置信度后ASD的风险基因为47个(图1)。
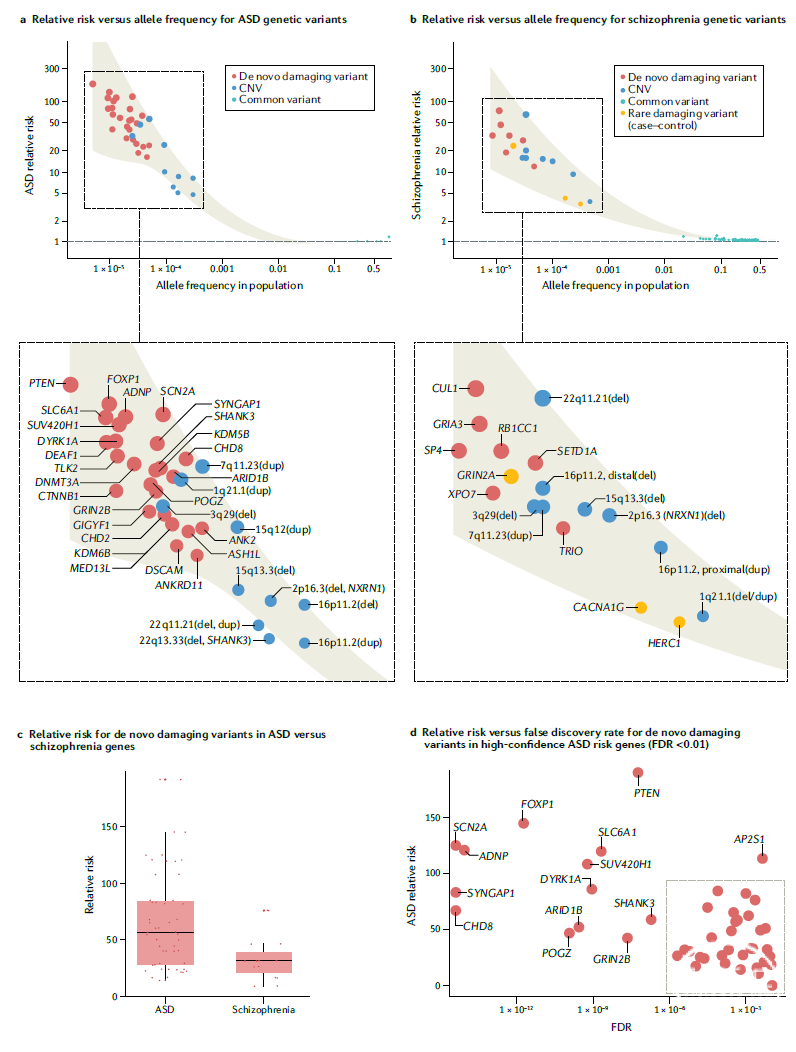
图1
交叉神经科学发展
上述基因组分析揭示了生物学的多样性和基因组的高度异质性,人们担心单一基因突变,以及针对于突变进行的相关治疗,可能只适用于特定突变的ASD患者,而不具有普适性。为此,在过去十年中,交叉神经科学领域得到迅速发展,其通过分子水平、细胞水平和环路水平以及跨多个维度(包括解剖定位和发育时间)对不同 ASD风险基因的重叠或交叉分析,旨在阐明ASD病理学的核心特征。
多效性问题
将在ASD患者中找到的风险基因转化为对病理生物学机制的理解以及寻找相关治疗靶点,面临的主要挑战之一是基因的多效性,因为它们在整个大脑发育和功能中参与不同角色,取决于细胞类型、大脑区域和/或发育阶段等。
因此,尽管某个明确的ASD风险基因可能在生物学上看似合理的过程中有一个被充分研究和描述的角色,但这并不一定意味着这是该基因发挥的唯一功能,或者它一定是促成ASD出现的角色。此外,由于物种差异的存在,依赖于动物模型的研究为人类神经发育障碍性疾病研究带来了一定的阻碍,这也极大地推动了对ASD风险基因的交叉神经科学的发展。
不完整的基因本体论
交叉神经科学方法基于这样的假设:特定疾病(如 ASD)的风险基因子集将指向从基因到复杂行为之间某个共同的异常交叉点。这种逻辑反映在ASD最早的测序研究中,这些基因存在功能上的重叠,例如共同影响突触的结构和功能、染色质修饰和转录调控等。
基于大量的靶向测序研究鉴定了第一批与ASD相关的编码突触蛋白质的基因,包括NRXN1、SHANK3、NLGN3和NLGN4等。随着ASD风险基因库的不断扩充,还发现了与染色质修饰和转录调控有关的基因趋同证据。然而,这些基于现有数据聚类的基因本体论分析,可能会倾向于更常见、更易研究的功能类别、细胞类型和发育时间点,从而导致偏移的产生,得到不完整的生物学解释。
大脑基因表达数据库
随着对ASD风险基因认识的不断增加,目前已识别出与ASD密切相关的大脑发育的解剖区域、细胞类型和/或发育阶段。
这些研究基于两个基本的假设:首先,这些基因存在高度协调的时空表达重叠性;其次,在正常大脑发育的背景下识别一组不同的ASD风险基因之间的重叠点是一种与核心病理生物学密切相关的三角参数方法。
在这方面最早的研究使用基于网络的方法来分析来自人类大脑发育轨迹的数据(BrainSpan Atlas of the Developing Human Brain),分析了正常人类大脑的多个解剖区域的从早期妊娠到成年晚期阶段的转录组数据。此外,目前已经进行了大量关于人类基因表达数据库与ASD风险基因相关的交互性分析(图2)。
比较尸检研究
识别ASD风险基因的交叉性的另一种方法是比较来自不同ASD个体和正常人死后脑组织的分子表达谱。
最早的研究分析了ASD患者包括前脑皮层、颞叶皮层和小脑等部位在内的基因转录情况,发现ASD患者的额叶和颞叶区域表达较正常组减弱。这一结果在随后进行的更大规模的队列研究中得到验证。尸检研究还发现,与对照组相比,ASD患者的大脑中神经元和突触相关基因的下调以及小胶质细胞和免疫相关基因的上调(图2)。

图2
患者细胞来源的类脑系统
对来自人类诱导多能干细胞(iPSC)分化的单层细胞模型以及人类大脑发育的3D模型(也称为类器官)进行了功能和转录分析。
在过去的几年里,有研究比较了来自ASD患者的 iPSC与正常个体的 iPSC 产生的神经元。与对照组相比,ASD和大头畸形患者iPSC分化的神经元出现共同的特征包括细胞增殖增加、神经元分化加速、突触发育受损以及自发和同步神经元活动减少等表型。
随着类器官技术的不断发展,这将是一个非常有前途的研究方法,可以更细致地了解早期神经发育过程中发生的分子水平和细胞类型水平的变化,包括与ASD相关的变化。
遗传风险的平行建模
对ASD最早出现病理改变的时间和部位的认识为潜在生物学研究以及平行研究ASD风险基因提供了机会,包括细胞类型和感兴趣的发育阶段。近年来, CRISPR技术的进步促进了体外和体内对多个ASD相关基因的平行研究,从而明确基因功能的交叉点。
最初的体外研究揭示了十几个ASD风险基因对iPSC或神经元前体细胞来源的神经元的影响。这些研究表明,ASD风险基因异常导致神经元分化受损和自发突触活动减少,表明多个ASD风险基因导致同一功能异常改变的重叠性。未来,合并CRISPR筛选技术的进步,能够对大量基因进行功能筛选,包括细胞增殖、分化以及在单细胞层面的基因表达和染色质可及性分析。目前,患者衍生的iPSC模型系统存在不同患者来源细胞的个体差异性和细胞群的异质性,以及小样本量等因素的影响。
总结
本文系统地综述了当前在基因组学和交叉神经科学方面对ASD疾病的研究进展,为阐明ASD的病理生物学特征和寻找潜在的治疗提供了方向。
AiBrain内容团队为大家整理了所有文章的pdf,如有需要,请公众号后台留言“pdf”或于文末添加AiBrain助手微信获取。
文献PMID:35440779
文献DOI:
10.1038/s41583-022-00576-7

往期精彩回顾✦






声明:脑医汇旗下神外资讯、神介资讯、脑医咨询、AiBrain所发表内容之知识产权为脑医汇及主办方、原作者等相关权利人所有。未经许可,禁止进行转载、摘编、复制、裁切、录制等。经许可授权使用,亦须注明来源。欢迎转发、分享
