病史简介
患者,女,45岁,因“左手抽动伴头晕1年余,头痛5天”入院。
患者1年余前出现左手抽动伴头晕不适,持续约4-5分钟好转,1月余前出现全身肢体抽搐不适,伴意识丧失,持续1-2分钟好转,5天前出现剧烈头痛,难忍,伴恶心呕吐,当地医院CT示:右额占位。予对症支持治疗,头痛好转,今为进一步诊治来我院就诊。
头颅MR增强扫描示:右侧额叶胶质瘤累及胼胝体及左侧额叶,少突胶质瘤首先考虑(图1)。
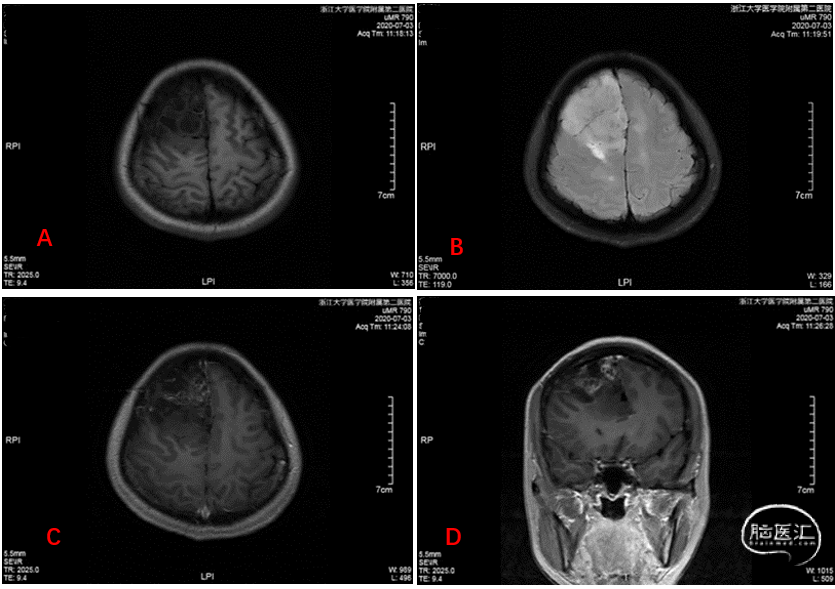
图1. 右侧额叶肿块,累及胼胝体及左侧额叶。A: 长T1信号;B:长T2信号;C:T1+C横断位显示右额叶肿块不均匀强化;D:T1+C冠状位增强扫描显示肿块不均匀强化。
入院后完善相关检查,排除手术禁忌后行“右侧额叶大脑病损切除术”,术中见肿瘤组织质韧、鱼肉样、血供中等、体积大、灰红色;边界不清,肿瘤与周边脑组织粘连,深部达胼胝体及侧脑室边,后方接近功能区,予全切除肿瘤。
病理诊断
大体检查:灰白灰红组织一堆,总大小6*5*1.5cm,质中。
显微镜观察:低倍镜下肿瘤呈弥漫性浸润性生长,细胞密度中等,间质粘液样变性(图2A),局部可见胞浆丰富核偏位的小肥胖型细胞(图2B),局部小血管内皮增生(图2C),伴见钙化(图2D);高倍镜细胞形态较一致,核增大,圆形卵圆形,染色质粗,部分呈胡椒盐样,周边细胞密度低的区域可见煎蛋样细胞(核周空晕),核分裂偶见。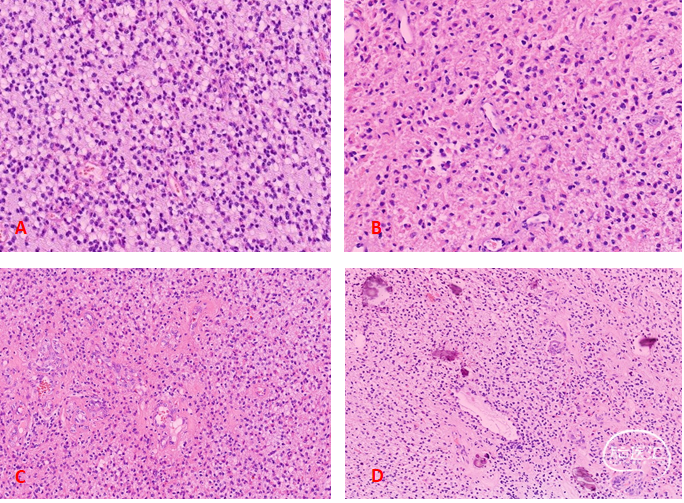
图2. A:少突胶质瘤细胞密度高,核圆形卵圆形,间质粘液样变;B:部分区域可见胞浆丰富嗜酸性,核偏位的小肥胖型细胞;C:微血管内皮细胞增生;D:部分区域可见钙化。免疫组化结果:GFAP+,Olig-2+,Nestin-,Syn部分-,IDH1(H09)+,ATRX存在,CD34血管+,BRAF-,Ki-67约15%+(图3)。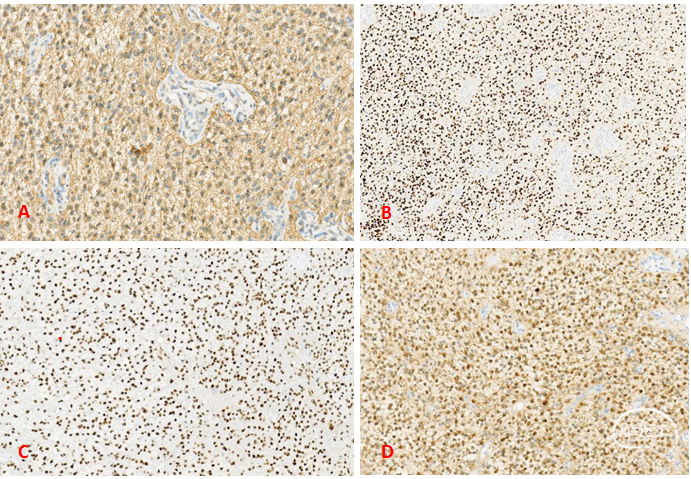
图3. 免疫组化标记表达。A:GFAP弥漫阳性;B:Olig-2核表达;C:ATRX存在;D:IDH(H09)弥漫阳性。分子检测(图4-6):符合1p/19q杂合性共缺失(FISH);IDH1外显子4突变阳性(R132H)(SANGER测序法);IDH2外显子4野生型(SANGER测序法);MGMT启动子甲基化(MSP-荧光探针法);TERT启动子突变阳性(C250T)(SANGER测序法);CDKN2A基因纯合性缺失(FISH)。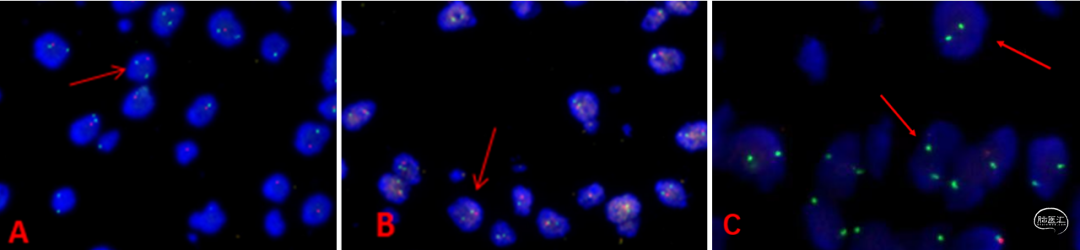
图4. A:1p杂合性缺失(红色箭头所示);B:19q杂合性缺失(红色箭头所示);C:CDKN2A纯合性缺失(红色箭头所示)。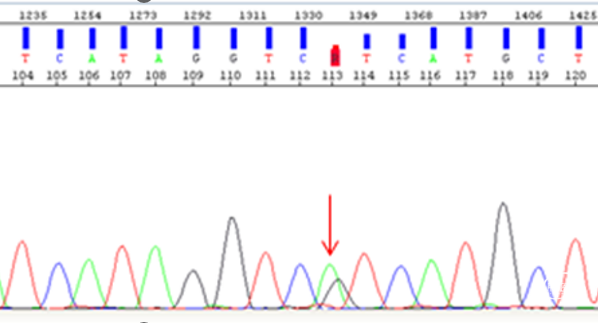
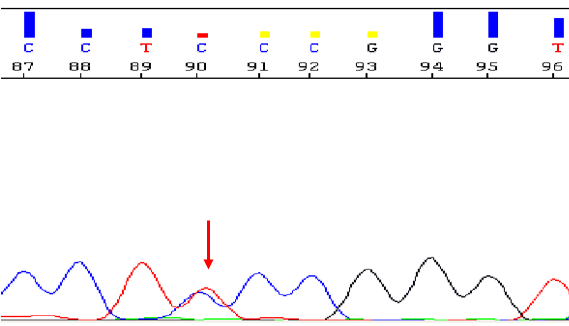
图6. 红色箭头所示TERT C250T启动子突变。最终诊断:少突胶质瘤,IDH突变和1p/19q共缺失,CNS WHO 3级。讨论
少突胶质瘤,IDH突变和1p/19q共缺失是一种弥漫浸润性胶质瘤,伴有IDH1或IDH2突变,染色体臂1p和19q共缺失,CNS WHO 2或3级。好发于成人,儿童罕见;CNS WHO 2级平均年龄43岁,CNS WHO 3级平均年龄50岁;额叶多见(59%),约2/3患者主要症状为癫痫发作。
IDH突变和1p/19q共缺失型少突胶质瘤在CT上通常表现为低密度或等密度肿块,多位于皮质及皮质下白质。钙化常见。MRI典型表现为T1低信号和T2高信号肿块,肿瘤边缘不清楚。
IDH突变和1p/19q共缺失少突胶质瘤细胞起源尚不清楚,可能起源于少突胶质前体细胞[1]。少突胶质瘤定义是IDH1密码子132或IDH2密码子172的错义突变,合并1p和19q全臂的缺失。90%以上的少突胶质瘤IDH突变为经典的IDH1 p.R132H突变,其余为非经典型突变。绝大多数少突胶质瘤携带TERT启动子热点突变,但发生于青少年的往往缺乏TERT启动子突变[1]。TERT启动子突变被认为是少突胶质瘤发展过程中的早期(即克隆)事件,在肿瘤进展和复发时稳定存在。表观遗传学上,MGMT启动子甲基化可在大多数少突胶质瘤中检测到[2]。
大体上少突胶质瘤表现为位于皮质和白质内的一个边界相对清楚、柔软、灰粉色的肿块。局灶可见软脑膜浸润。钙化常见,并可有砂质结构。囊性退行性变以及瘤内出血也是常见的。少见粘液样变性呈凝胶状。在CNS WHO 3级肿瘤中可以看到坏死区域。组织学上,典型的少突胶质细胞具有一致的圆形核,比正常的少突胶质细胞略大,染色质加深或呈细颗粒的胡椒盐状。核膜通常明显。HE表现为典型的蜂窝状或煎蛋状的外观,这是一个重要的诊断特征。少突胶质瘤可含有胶质纤维少突细胞和小肥胖细胞[3]。胶质纤维少突细胞和小肥胖细胞在CNS WHO 3级肿瘤中较为常见。本例肿瘤细胞主要以胶质纤维少突细胞为主(图2A),局部区域可见小肥胖细胞(图2B)。偶尔CNS WHO 3级少突胶质瘤以多核巨细胞为特征,极少数病例含有肉瘤区域[4,5]。肿瘤内和肿瘤周围组织常见微钙化(图2D)。少突胶质瘤典型的表现为密集的毛细血管分支网,类似于细铁丝网。在CNS WHO 3级肿瘤中,可以有局灶性或弥漫性病理性微血管增生(图2C)。CNS WHO 2级少突胶质瘤的有丝分裂活性低或不存在,但CNS WHO 3级肿瘤的有丝分裂活性通常显著。
关于WHO CNS分级,WHO将IDH突变和1p/19q共缺失的少突胶质瘤分为CNS WHO 2级和CNS WHO3级,但目前分级标准尚未明确。与高级别相关的组织学特征是细胞丰富、明显的细胞异型性、有丝分裂活跃、病理性微血管增生和伴或不伴栅栏状的坏死。CNS WHO 3级少突胶质瘤通常具有以上几种特征。在一项组织学定义的少突胶质瘤研究中,微血管增生和活跃的有丝分裂活动≥2.5个有丝分裂/mm²(相当于6个有丝分裂/10 HPF,直径0.55 mm,面积0.24mm²),认为是生存期短的指标[6]。在手术切除标本中检测到罕见的有丝分裂并不足以诊断CNS WHO 3级。关于Ki-67(MIB1)阈值,由于不同机构计数方法不统一,染色结果存在显著差异,因此亦尚未确定。一项研究报道,CNS WHO 3级少突胶质瘤患者的Ki-67指数≥15%,是生存期较短的独立标记[7]。CDKN2A和/或CDKN2B纯合性缺失在一小部分(<10%)CNS WHO 3级少突胶质瘤报道,尚未在CNS WHO 2级少突胶质瘤中发现,且与低生存率有关,与微血管增生及坏死无关[2]。因此,CDKN2A/B纯合性缺失与少突胶质瘤CNS WHO 3级密切相关。
免疫组化大多数少突胶质瘤表现出IDH1 p.R132H 阳性反应,ATRX核表达,通常缺乏广泛的P53核染色,GFAP可在混合反应的星形胶质细胞中检测到,但也可染色肿瘤细胞。少突胶质细胞谱系转录因子OLIG1、OLIG2和SOX10在少突胶质瘤中表达。在一项研究中,在88.5%的CNS WHO 3级少突胶质瘤中发现了α-internexin蛋白的免疫染色,但不认为是1p/19q共缺失的替代标记[8]。
IDH突变和1p/19q共缺失少突胶质瘤有良好的治疗反应,中位生存期>10年。CNS WHO 3级少突胶质瘤患者联合放疗和丙卡嗪、洛莫司汀和长春新碱治疗(PCV)化疗显示中位生存期为14年[9]。少突胶质瘤通常局部复发,可表现为软脑膜播散,复发时发生恶性进展常见。
总结:本例是伴CDKN2A纯合性缺失的CNS WHO 3级少突胶质瘤。虽然CNS WHO 3级的IDH突变和1p/19q共缺失少突胶质瘤诊断级别标准目前还未明确。但细胞丰富、明显的细胞异型性、有丝分裂活跃、病理微血管增生和伴或不伴栅栏状的坏死等是CNS WHO 3级的主要组织学特点,而且CDKN2A/B纯合性缺失在IDH突变和1p/19q共缺失少突胶质瘤中的意义值得关注。参考文献
1. Persson, A.I., et al., Non-stem cell origin for oligodendroglioma. Cancer Cell, 2010. 18(6): p. 669-82.2. Appay, R., et al., CDKN2A homozygous deletion is a strong adverse prognosis factor in diffuse malignant IDH-mutant gliomas. Neuro Oncol, 2019. 21(12): p. 1519-1528.3. Herpers, M.J. and H. Budka, Glial fibrillary acidic protein (GFAP) in oligodendroglial tumors: gliofibrillary oligodendroglioma and transitional oligoastrocytoma as subtypes of oligodendroglioma. Acta Neuropathol, 1984. 64(4): p. 265-72.4. Cancer Genome Atlas Research, N., et al., Comprehensive, Integrative Genomic Analysis of Diffuse Lower-Grade Gliomas. N Engl J Med, 2015. 372(26): p. 2481-98.5. Suzuki, H., et al., Mutational landscape and clonal architecture in grade II and III gliomas. Nat Genet, 2015. 47(5): p. 458-68.6. Giannini, C., et al., Oligodendrogliomas: reproducibility and prognostic value of histologic diagnosis and grading. J Neuropathol Exp Neurol, 2001. 60(3): p. 248-62.7. Pouget, C., et al., Ki-67 and MCM6 labeling indices are correlated with overall survival in anaplastic oligodendroglioma, IDH1-mutant and 1p/19q-codeleted: a multicenter study from the French POLA network. Brain Pathol, 2020. 30(3): p. 465-478.8. Figarella-Branger, D., et al., Mitotic index, microvascular proliferation, and necrosis define 3 groups of 1p/19q codeleted anaplastic oligodendrogliomas associated with different genomic alterations. Neuro Oncol, 2014. 16(9): p. 1244-54.9. van den Bent, M.J., et al., Diffuse Infiltrating Oligodendroglioma and Astrocytoma. J Clin Oncol, 2017. 35(21): p. 2394-2401.
(本文由浙二神外周刊原创,浙江大学医学院附属第二医院病理科李雁青主治医师整理,许晶虹副主任医师审校,张建民主任医师终审)
声明:脑医汇旗下神外资讯、神介资讯、脑医咨询、AiBrain所发表内容之知识产权为脑医汇及主办方、原作者等相关权利人所有。未经许可,禁止进行转载、摘编、复制、裁切、录制等。经许可授权使用,亦须注明来源。欢迎转发、分享。



