



提示
前言
神经病理是临床诊断,尤其是神经肿瘤诊断的最重要一环。根据神经病理的最终诊断结果决定下一步治疗方案。近年来,随着分子病理及基因检测的不断开展和深入,中枢神经系统病理诊断的更新也越来越快。如2021年底出版的第5版CNS WHO分类,增加了22种肿瘤新类型,修订了13种肿瘤的命名,其主要变化是分子诊断的权重更大,强调整合诊断和分层报告的重要性。因此,需要我们不断学习,与时俱进,提高诊断水平,以便更好地治疗病人。我院病理科自2011年起成立中枢神经病理亚专科,现有亚专科组成员8名,其中副主任医师3名,主治医师2名,住院医师3名。已开展神经病理相关的组织学、特殊染色、免疫组织化学及分子病理学(FISH、测序、甲基化检测等)。借助长期的临床实践,并于2011年和美国UCLA神经病理团队建立了联合会诊平台,不断交流合作,积累了丰富的经验。从第339期开始我们报道由浙大二院病理科神经病理团队汇总的系列病例,以期和大家充分交流讨论。欢迎大家批评指正。
病史简介
患者,女,58岁,因“走路不稳伴尿失禁半年余”入院。
患者半年余前出现走路不稳,憋尿时有尿失禁。1月余前查头颅MRI平扫示:松果体区占位。为进一步诊治来我院,门诊拟“松果体区肿瘤”收住入院。
查体:双侧瞳孔等大等圆,直径3mm,对光反射灵敏,左眼球活动受限,四肢肌力5级,肌张力正常,病理征阴性,左侧跟膝胫试验阳性。
我院头颅磁共振增强扫描:松果体区占位性病变,考虑肿瘤,生殖细胞瘤较脑膜瘤可能。两侧侧脑室及第三脑室扩大(图1)。
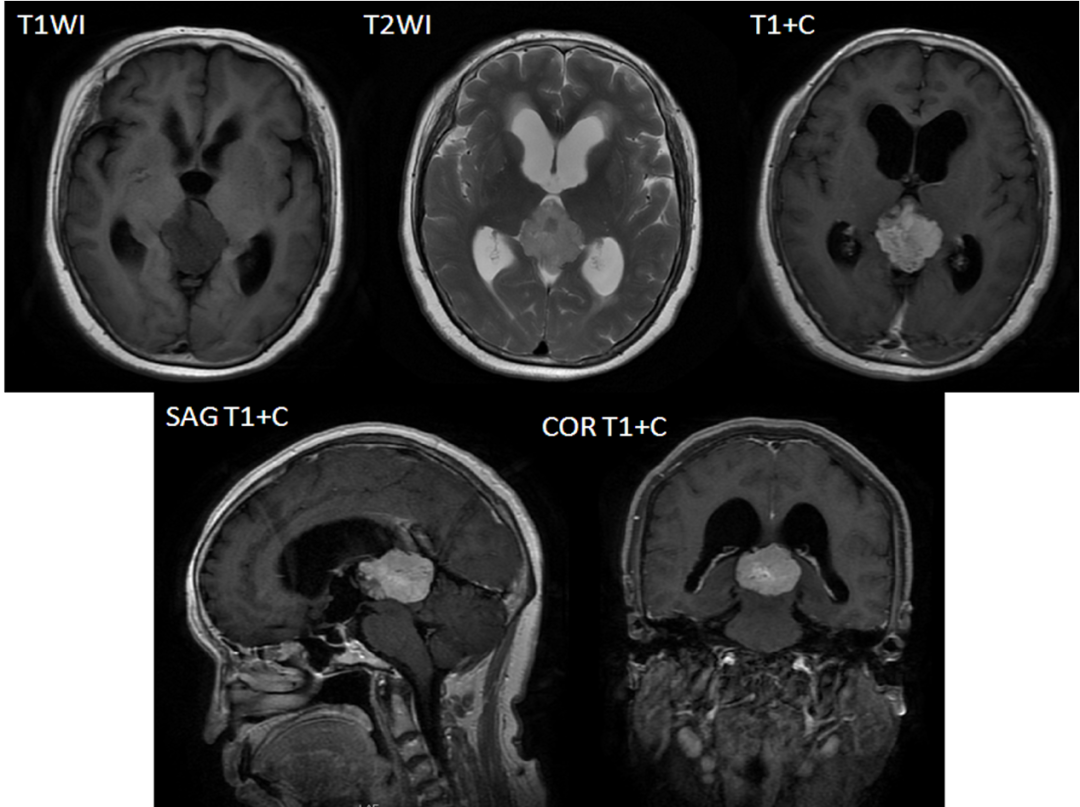
图1. 所示松果体区见不规则异常信号灶,大小约3.7*3.3cm,T1WI低信号、T2WI略高信号,增强扫描可见明显强化,两侧侧脑室及第三脑室扩大;余各层脑实质内未见异常信号影,脑池和脑沟形态及位置无殊,中线结构无移位。
诊治经过
患者入院后完善相关术前检查,排除手术禁忌后行开颅松果体区肿瘤切除术。术中见肿瘤,呈红色,血供一般,周围包膜完整,界清,质软。
病理诊断
大体检查:(松果体区)送检破碎灰白色组织一堆,大小4.8*2.5*1.8cm。
组织学观察:肿瘤表面被有纤维包膜,内由纤维血管间隔呈假小叶结构,局部可见微囊形成(图2. A、B)。肿瘤细胞密度中等,主要有两种形态学表现:体积较小、形态较一致的肿瘤细胞聚集成团,呈少突细胞样,核圆形至卵圆形,胞浆较少、粉染至透亮,染色质粗糙颗粒状(图2. C、D)。体积较大的神经节细胞样肿瘤细胞散在分布其中,胞浆丰富、嗜伊红(图2. E);部分细胞多形性明显,核大、不规则、深染,核仁明显,亦可见双核和多核细胞(图2. F)。未见核分裂及坏死。
免疫组化结果:Syn弥漫强+,NF +,CgA灶性点状+,NeuN散在+,GFAP和S-100示反应性星形细胞,EMA -,CAM5.2 -,CK(AE1/AE3)-,CD99 -,P53 -,CD68散在+,CD34示血管,Ki-67增殖指数约2-5%(图3)。

图2. 肿瘤界限清楚(A),呈假小叶结构(B);肿瘤细胞密度中等(C),见少突细胞样细胞(D)和神经节细胞样细胞(E),部分细胞多形性显著(F)。

图3. 肿瘤Syn弥漫强阳性,CgA与NF表达, NeuN示神经节细胞样细胞。
最终诊断:中分化松果体实质肿瘤(PPTID),多形性亚型,CNS WHO 2级。
讨论
原发于松果体区域的肿瘤很少见,占39岁以下人群原发性脑和中枢神经系统肿瘤的1.2%-3.2%[1]。包括来源于松果体细胞的肿瘤:松果体细胞瘤(CNS WHO 1),中分化松果体实质肿瘤(CNS WHO 2-3级),松果体母细胞瘤(CNS WHO 4级);可能来源于下联合器特化的室管膜细胞的松果体区乳头状肿瘤(CNS WHO 2-3级);以及第五版WHO中枢神经系统肿瘤分类中新提出的松果体区促结缔组织增生性黏液样肿瘤,SMARCB1-突变型[2]。
中分化松果体实质肿瘤(Pineal parenchymal tumour of intermediate differentiation, PPTID)由形态较一致的圆形细胞呈弥漫片状或分叶状排列。肿瘤的组织学发生与松果体细胞相关。PPTID分化程度介于松果体细胞瘤与松果体母细胞瘤之间,中度恶性。有局部复发和脑脊髓播散的潜能。
PPTID约占所有松果体实质瘤的45%,中位发病年龄33岁(3.5-64岁),女性稍多见,男女比例约0.8:1。
PPTID临床可表现为由中脑导水管堵塞引起的颅内压增高、神经-眼功能障碍(帕里诺综合征)、以及脑干或小脑功能障碍。
影像学上显示为球形、界清的肿块,常伴局部侵袭表现。
大体上,PPTID呈灰褐、界清肿块,切面均匀或颗粒状,可伴出血或囊性变。
组织学上,PPTID可表现为两种结构模式:弥漫型(神经细胞瘤样或少突胶质细胞瘤样)和/或分叶型(血管分隔的模糊小叶)。细胞密度中至高等,少至中等量胞浆,细胞核圆形、轻至中度异型性,染色质呈胡椒盐样。
松果体实质肿瘤的多形性亚型,可出现在松果体细胞瘤和低级别PPTID,呈多形性巨细胞或神经节样肿瘤细胞,呈畸形深染核。回顾性研究表明这些肿瘤细胞的多形性与恶性程度无关,但最终的病理诊断应结合其增殖指数考虑[3-5]。
PPTID通常表达Syn、NF和CgA。显示广泛的神经元分化。典型者弥漫性(> 50%细胞)核表达CRX,为参与视网膜和松果体分化的光感受器特异性转录因子[6]。
2019年,利用全外显子基因测序,Julieann C. Lee等人首先报道了3例PPTID中均有KBTBD4基因内的小片段插入(p.R313delinsPRR)。KBTBD4位于11p11.2,包含BTB和Kelch重复结构域,可调控基于Cullin3支架蛋白的E3泛素连接酶复合物[7]。之后3篇报道显示66.7%(6/9)、74% (20/27 )、16/16(100%)的PPTID存在KBTBD4的插入突变[8-10]。据此,KBTBD4的插入突变可为PPTID的诊断提供强有力的证据。此外,类似的KBTBD4插入突变还在非WNT/非SHH髓母细胞瘤报道,在肿瘤的表观遗传重编程中起驱动作用[11]。PPTID的基因拷贝数分布相对平坦。根据DNA甲基化可将PPTID分为两组:PPTID-A和PPTID-B,但其预后意义需要进一步评估[9,10]。
PPTID组织学上与中枢神经细胞瘤、少突胶质细胞瘤有相似,需鉴别。中枢神经细胞瘤不表达CgA,PPTID一般不表达NeuN;少突胶质细胞瘤OLIG2阳性、IDH突变和1p/19q共缺失。此外,结合影像学所示发病部位亦有提示意义。混合性松果细胞瘤-松果体母细胞瘤是由境界清楚的两种肿瘤组成,不应被诊断为PPTID。
在包括127例PPTID患者的29项研究中,中位总生存期为14年,5年总生存率为84%。中位无进展生存期为5.2年,5年无进展生存率为52%。肿瘤复发常有脊髓/软脑膜播散。
PPTID的细胞增殖指数低至中度,Ki-67指数为3.5%至16.1%。早在2000年,Jouvet等人提出,低级别(CNS WHO 2级)PPTID定义为低增殖活性(核分裂<6/10 HPF)伴NF的弥漫表达;高级别PPTID的定义是低增殖活性但无或仅有少数细胞表达NF,或高增殖活性(核分裂≥6/10 HPF)但多数细胞表达NF[12]。低级别PPTID患者5年总生存率为74%,复发率为26%;高级别PPTID患者5年总生存率为39%,复发率为56%,28%可发生脊髓播散[13]。在另一项研究中,低级别PPTID(Ki-67增殖指数<5%)的患者比高级别 PPTID(Ki-67增殖指数≥5%)患者的总生存期和无进展生存期更长[14]。综上,核分裂数、NF免疫阳性和Ki-67增殖指数的预后作用需要进一步研究,而分子亚群的预后作用仍有待确定。目前尚没有推荐的PPTID分级标准。
总结:本例为发生在成人的松果体实质肿瘤,伴细胞多形性与神经元分化。因其中一定数量的多形性细胞,本例还应与多形性黄色星形细胞瘤、多形性星形细胞瘤相鉴别。KBTBD4基因检测可帮助明确诊断。
参考文献
1. Ostrom, Q.T., et al., CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2011-2015. Neuro Oncol, 2018. 20(suppl_4): p. iv1-iv86.
2. Thomas, C., et al., Desmoplastic myxoid tumor, SMARCB1-mutant: clinical, histopathological and molecular characterization of a pineal region tumor encountered in adolescents and adults. Acta Neuropathol, 2020. 139(2): p. 277-286.
3. Fevre-Montange, M., et al., Pineocytoma and pineal parenchymal tumors of intermediate differentiation presenting cytologic pleomorphism: a multicenter study. Brain Pathol, 2008. 18(3): p. 354-9.
4. Ito, T., et al., [Pineal Parenchymal Tumor with Marked Cytologic Pleomorphism: Is there a Correlation with the Malignancy Grade?]. No Shinkei Geka, 2016. 44(6): p. 481-7.
5. Sasaki, A., K. Horiguchi, and Y. Nakazato, Pineal parenchymal tumor of intermediate differentiation with cytologic pleomorphism. Neuropathology, 2006. 26(3): p. 212-7.
6. Coy, S., et al., Nuclear CRX and FOXJ1 Expression Differentiates Non-Germ Cell Pineal Region Tumors and Supports the Ependymal Differentiation of Papillary Tumor of the Pineal Region. Am J Surg Pathol, 2017. 41(10): p. 1410-1421.
7. Lee, J.C., et al., Recurrent KBTBD4 small in-frame insertions and absence of DROSHA deletion or DICER1 mutation differentiate pineal parenchymal tumor of intermediate differentiation (PPTID) from pineoblastoma. Acta Neuropathol, 2019. 137(5): p. 851-854.
8. Uchida, E., et al., Role of proliferative marker index and KBTBD4 mutation in the pathological diagnosis of pineal parenchymal tumors. Brain Tumor Pathol, 2022.
9. Liu, A.P.Y., et al., Clinical and molecular heterogeneity of pineal parenchymal tumors: a consensus study. Acta Neuropathol, 2021. 141(5): p. 771-785.
10. Pfaff, E., et al., Molecular subgrouping of primary pineal parenchymal tumors reveals distinct subtypes correlated with clinical parameters and genetic alterations. Acta Neuropathol, 2020. 139(2): p. 243-257.
11. Chen, Z., et al., Disease-associated KBTBD4 mutations in medulloblastoma elicit neomorphic ubiquitylation activity to promote CoREST degradation. Cell Death Differ, 2022.
12. Jouvet, A., et al., Pineal parenchymal tumors: a correlation of histological features with prognosis in 66 cases. Brain Pathol, 2000. 10(1): p. 49-60.
13. Fauchon, F., et al., Parenchymal pineal tumors: a clinicopathological study of 76 cases. Int J Radiat Oncol Biol Phys, 2000. 46(4): p. 959-68.
14. Yu, T., et al., Twenty-seven cases of pineal parenchymal tumours of intermediate differentiation: mitotic count, Ki-67 labelling index and extent of resection predict prognosis. J Neurol Neurosurg Psychiatry, 2016. 87(4): p. 386-95.
