


《Journal of Clinical Medicine》杂志2019年11月12日在线发表日本东京Toranomon Hospital的Hiroshi Nishioka和Moriyama Neurological Center Hospital的Shozo Yamada撰写的长篇综述《库欣病Cushing’s Disease》(doi:10.3390/jcm8111951)。

对于库欣病(CD)患者,及时的诊断和治疗是至关重要的,有利于长期结果,尽管这仍然是一个具有挑战性的任务。对于某些患者,即使是采用有有序地逐步诊断方法,库欣病的鉴别诊断仍然存在困难。此外,尽管使用高分辨率磁共振成像(MRI)结合先进的优良的序列,有些肿瘤仍然是不可见的。手术,使用各种手术方法以安全地最大限度地切除肿瘤,仍然是大多数库欣病患者的首选治疗方法。持续的或手术失败后复发的库欣病需要进一步治疗,包括重复手术、药物治疗、放疗,有时也会行双侧肾上腺切除术。这些治疗方法有各自的优势和劣势。然而,最重要的是,这种复杂的疾病应该由互相协作的专家组成的多学科团队管理。此外,个性化和而基于个体的方法是最重要的,以达到高的成功率,同时尽量减少副反应的发生,提高患者的生存质量。最后,高度期待最近在分子水平上对库欣病病理生理机制的深入了解,将在不久的将来给这个可怕的疾病带来更准确的诊断检测和有效的治疗。
第一节 介绍
1932年,Harvey W. Cushing报告了12例有严重代谢紊乱的患者,他将其归因于垂体嗜碱性肿瘤。目前,库欣综合征(CS)指的是血浆糖皮质激素水平异常升高的临床症状和体征。外源性(医源性)库欣综合征(CS)常见,而内源性库欣综合征(CS)少见,可概括性地分为促肾上腺皮质激素(ACTH)依赖性(约80%);库欣病(CD)、异位分泌ACTH的肿瘤、分泌促肾上腺皮质激素释放激素(CRH)肿瘤、以及与ACTH无关(20%);肾上腺肿瘤、垂体癌和肾上腺大结节增生(AIMAH)。库欣病是一种由垂体促肾上腺皮质激素细胞肿瘤引起的罕见疾病,是内源性库欣综合征(CS)最常见的病因(成人中约70%)。垂体肿瘤过度分泌ACTH,缺乏下丘脑-垂体-肾上腺轴反馈调节;因此,肾上腺分泌超量的皮质醇。长期全身暴露于升高的皮质醇水平,由于合并症、死亡率增加和健康相关生存质量(HRQoL)受损,会导致库欣病患者的临床负担加重,最近的研究清楚地证明,早期准确的诊断和治疗对长期结果的影响[2 7]。然而,尽管有显著进展,库欣病的诊断和治疗仍然具有挑战。
第二节 流行病学
虽然关于库欣病的详细的流行病学数据有限,但根据几项以人群为基础的研究,该病的患病率估计为近40 例/ 100万,发病率为每年每百万人1.2-2.4 例。由于存在轻度和/或非典型症状而未被认识的患者和循环型库欣患者,其患病率可能被低估。
在成人中,女性患库欣病的频率是男性的3倍,症状通常出现在30岁至60岁之间。据报道,库欣病在较年轻的年龄出现,男性的临床表现比女性的更严重。在老年人中,男性和女性之间的库欣病患病率差异不大。
大约75%-90%的儿童库欣综合征(CS)病例是由库欣病引起的。库欣病在6岁以下的儿童中并不常见;库欣综合征(CS)的肾上腺病因是低龄儿童的典型病因。与成人患者一样,儿童和青少年库欣综合征(CS)患者总体上以女性比男性占优势,并且随着年龄越小而越少。
最近Wengander等人提出,库欣综合征(CS)患者异位ACTH综合征的比例高于既往的报道。在研究的库欣综合征(CS)患者中,约有一半是由库欣病引起的,四分之一是由异位性分泌ACTH的肿瘤引起的,四分之一是由肾上腺疾病引起的。相反,Hirsch等人报告,可能是因为与轻度皮质醇增多症相关的分泌促肾上腺皮质激素的偶发瘤的检出增加,库欣综合征(CS)的肾上腺病因的相对比例正在上升。
第三节 分子病理生理学
3.1遗传易感性
引起库欣病的垂体促肾上腺皮质激素细胞肿瘤多为散发性,只有少数病例涉及多种遗传内分泌综合征。这些遗传内分泌综合征包括家族性孤立性垂体瘤(FIPA;AIP)、多发内分泌肿瘤1型(MEN1)和4型(CDKN1B)、Carney复合征(PRKAR1A)、DICER1综合征(DICER1)。促肾上腺皮质激素细胞肿瘤约占家族性孤立性垂体瘤(FIPA)中所有垂体肿瘤的5%。迄今为止,在库欣病中还没有GNAS或PRKAR1A的生殖细胞系突变的报道。生殖细胞系DICER1突变在垂体母细胞瘤中已有报道,而垂体母细胞瘤是一种罕见的婴儿起病的库欣病的病因。
3.2.基因档案
糖皮质激素肿瘤的发病机制仍不清楚。最近的一个重要进展是在大约23-60%的功能性促肾上腺皮质激素细胞肿瘤中检测到泛素蛋白特异性肽酶(ubiquitin-specific peptidase)8 (USP8)的基因突变(表1)。体细胞突变是促肾上腺皮质激素细胞肿瘤特定的,会导致表皮生长因子受体(EGFR)表达的增加和激活阿黑皮素原(proopiomelanocortin ,POMC)基因转录。典型的USP8突变的表型表现为中年妇女的库欣病伴有小肿瘤。这种突变在Crooke细胞肿瘤中并不常见,是促肾上腺皮质激素细胞肿瘤的一种组织学亚型,常表现出进袭性的临床行为。具有USP8突变的促肾上腺皮质激素细胞肿瘤的SSTR5和MGMT表达水平明显高于野生型。因此,与野生型肿瘤相比,有USP8突变的肿瘤患者可能有更好的手术结果,并且对靶向SSTR5的生长抑素类似物的反应可能更好。然而,最近的一项研究报道,有USP8突变的肿瘤患者术后复发更早、更频繁。Faucz等人发现,几乎三分之一(31%)的儿科库欣病患者的肿瘤中存在体细胞USP8突变。患有USP8突变的儿科患者的总体疾病更严重,首要的(primary)手术切除的失败率更高,复发的风险也更高。
表10.报道的库欣病患者泛素蛋白特异性肽酶8 (USP8)突变频率及其临床特征。

最近有报道称,对与USP8突变相关的库欣病研究的回顾性调查中发现,研究系列中的生化结果和手术结果存在异质性。Wanichi等人提出,进一步的多中心前瞻性研究将对这些库欣病患者促肾上腺皮质激素细胞肿瘤提供关于表型、治疗反应和预后影响的更一致的信息。
Sesta等人的研究表明,带有USP8突变的促肾上腺皮质激素细胞肿瘤呈现更典型的促肾上腺皮质激素表型,并且与蛋白质降解相关的几个基因的表达减少。另一方面,USP8突变发生在功能性和非功能性促皮质激素肿瘤细胞中。Bujko等人的研究表明,USP8具有多效性(pleiotropic efffect),不仅局限于在不同途径中多种基因的EGFR信号转导和表达的水平。对可能成为治疗方法的潜在靶点USP8的蛋白靶点仍不清楚。
所有在库欣病中识别的USP8突变都是体细胞杂合的单点突变,除了最近的一个病例报告中描述了一例儿科库欣病患者的新生生殖系USP8突变。除了复发性库欣病外,该患者还表现为发育迟缓、异形特点和其他表现,这可能是一种新的遗传综合征的特征。
Chen等人报道,在携带野生型USP8的促肾上腺皮质激素细胞肿瘤中检测到USP48和BRAF V600E的频繁突变。最近,Sbiera等人发现USP48突变在USP8野生型肿瘤中较为频繁,并且以与sonic hedgehog信号通路活化相一致的方式增强CRH诱导的激素的产生。BRAF V600E变异在促肾上腺皮质激素细胞肿瘤中极为罕见,而TP53致病性变异比以前假设的更常见,尤其是在较大的肿瘤中。相反,HSP90的过表达参与了库欣病的形成,并导致部分糖皮质激素的抵抗性。
第四节临床特点(暂缺)
第五节诊断和鉴别诊断(暂缺)
第六节 死亡率
以前,与库欣病相关的死亡率很差;5年死亡率为50%。然而,目前的研究表明,库欣病患者的标准化死亡率(SMR)显著增加(范围:1.7-4.8)。根据一项大型队列研究,库欣综合征患者病死率(危险比[HR] 2.3)、静脉血栓栓塞(VTE)(HR 2.6)、心肌缺血性梗死(HR 3.7)、卒中(HR 2.0)、消化性溃疡(HR 2.0)、骨折(HR 1.4)和感染(HR 4.9)的死亡风险增加,甚至在诊断之前。肾上腺库欣综合征患者病死率 (HR2.4)和库欣病患者的(HR2.3)风险相似。一般来说,心血管疾病是最常见的死亡原因,其次是脑血管疾病、恶性肿瘤和感染性疾病。此外,最近有报道称自杀死亡率很高。以下的危险因素导致了SMR(标准死亡比)的增加:女性、老年、持续性高血压和糖代谢受损。
治疗后,持续性疾病患者、接受双侧肾上腺切除术患者和需要糖皮质激素替代治疗患者的亚组中,死亡率最高。
库欣病(CD)患者在“治愈”后的死亡风险的增加可能持续。根据Clayton等人的研究特别是在已缓解超过10年的库欣病患者中,由于循环系统疾病,,总体死亡风险比一般人群更高。皮质醇增多症总体持续时间和暴露程度是影响死亡率持续升高的重要因素。Lambert等人的研究表明,接受过治疗的库欣病患者死亡的预测因素包括暴露于超量糖皮质激素的持续时间、术前ACTH浓度和诊断时年龄较大。Osswald等人最近的一项研究表明,与诊断时的血清皮质醇峰值相比,暴露于超量的糖皮质激素的时间更能预测长期的精神疾病患病率和生存质量。因此,为了缩短暴露于活跃的库欣综合征的时间,及时诊断和有效治疗是非常必要的。
在各种治疗达到缓解的库欣病中,单纯垂体手术是首选的治疗,以确保最佳的结果。Clayton等人报告,虽然“已治愈”患者的总体死亡率有增加,但仅接受一次垂体手术的患者的总体死亡率是正常的。
第七节 治疗(暂缺)
第八节 库欣病的病理特点
8.1.组织学检查
第一步是确认所提交的组织是来自正常垂体还是垂体瘤。然后,完成基于细胞产生的激素的免疫组化分类,并最后 ,可以通过一些生物标志物和分子/遗传/表观遗传学研究来确定预后信息和治疗选项。库欣病手术中获得的组织样本通常很小,因为大多数库欣病的肿瘤是很小的微型肿瘤。相反,常提交的正常的垂体组织以确认不存在肿瘤。必须注意,尤其是当正常垂体组织显示弥漫性ACTH-阳性染色,要避免误诊为促肾上腺皮质激素细胞肿瘤或弥漫的促肾上腺皮质细胞增生。当从垂体中央部分获得小的组织样本时,那里的正常促肾上腺皮质细胞激素丰富,或来自嗜碱性侵袭的部分,可能会发生误诊。为了避免这种情况,银和网状蛋白染色(silver and reticulin stains)对显示网状蛋白纤维很重要。腺泡的网状结构在正常垂体组织中保留,但在肿瘤组织中丢失;此外,在增生的情况下可能会增大。
此外,在75-80%的各种病因引起的慢性皮质醇增多症患者中,由于胞质颗粒被均匀的玻璃样物质代替,正常垂体中的促肾上腺皮质激素细胞会发生变化。这些变化已知为Crooke玻璃样变,对它们的识别是至关重要的。存在这些变化与皮质醇增多症的程度和个体的易感性有关。这些变化涉及到在过度的糖皮质激素水平作用下,大量累积的细胞核周围的细胞角蛋白。这种累积可以用低分子量角蛋白(CAM5.2)免疫染色来显示,在细胞核周围呈牢固的(strong)环状,将PAS(过碘酸-Schiff反应)染色阳性颗粒和ACTH阳性颗粒转移至细胞外周。Crooke玻璃样变(Crooke’s hyaline changes)在没有皮质醇增多症的情况下是不存在的。因此,它们的存在可以被用来确认在垂体手术探查过程中没有库欣综合征征象的患者的库欣综合征(CS)。
相反,约20%的在手术中ACTH阳性的垂体瘤患者不显示Crooke变化,因此,缺乏Crooke变化并不总能证实不是库欣综合征。非肿瘤性地腺垂体促肾上腺皮质激素细胞的数目增加,被称为促肾上腺皮质激素细胞增生,在这种增生中,可能由于糖皮质激素的负反馈作用的下降或受到促肾上腺皮质激素释放激素(CRH)作用的增加,网状结构完整但延展。促肾上腺皮质激素细胞增生可能发生在原发性皮质机能减退(Addison病)或异位产生CRH的肿瘤。增生可以是局灶性的或是弥漫性的,且位于前叶的促肾上腺皮质激素细胞中。虽然不常见,促肾上腺皮质激素增生导致的库欣病已有报道。
肿瘤的分类是基于前叶激素免疫组化的结果。在缺乏明显的(distinct)激素含量的情况下,垂体转录因子可能有助于区分肿瘤细胞的分化;垂体特异性转录因子1 (Pit-1)、t-box转录因子(Tpit)、类固醇生成因子(SF-1)、雌激素受体α(ER-α)、GATA结合蛋白2 (GATA-2)都可被应用。其中,Tpit阳性被用来确认促肾上腺皮质激素细胞的来源。促肾上腺皮质激素细胞肿瘤可分为致密的和稀疏的颗粒型,这与电镜观察到的分泌颗粒模式有关。
8.1.1.致密颗粒型促肾上腺皮质激素细胞肿瘤
它们由嗜碱性细胞组成,在促肾上腺皮质激素和过碘酸-Schiff反应(PAS)染色中呈弥漫的和强烈的阳性(图11)。微肿瘤和小的大腺瘤(small macro-tumors)通常是这种类型的组织学。细胞电镜显示肿瘤由伸长的或角状的细胞组成,细胞器发育良好。分泌颗粒数量众多,在大小、形状和电子密度上有很大差异;中间丝(1型细丝)可呈成束的核周环状角蛋白细丝,CAM5.2免疫染色可见。
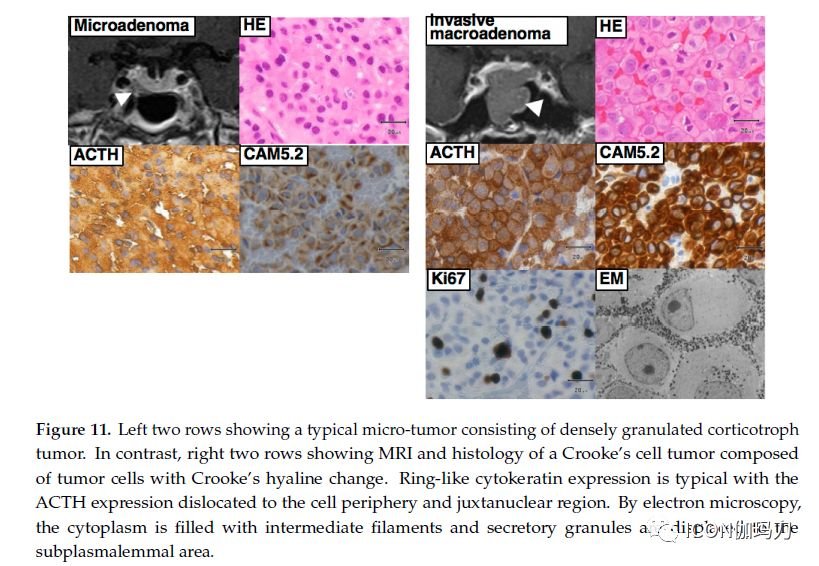
图11.左侧两行显示典型的微腺瘤,由致密颗粒型促肾上腺皮质激素细胞肿瘤组成。与此相反,右侧两行显示Crooke细胞肿瘤的MRI和组织学,该肿瘤由Crooke玻璃样变的肿瘤细胞组成。环状细胞角蛋白表达典型伴有ACTH表达移位至细胞外周和似核区域。通过电镜观察,胞质内充满中间丝,分泌颗粒被转移到质膜下区域。
8.1.2.稀疏颗粒型促肾上腺皮质激素细胞肿瘤
一些促肾上腺皮质激素细胞肿瘤是侵袭邻近组织的大腺瘤。在如此大的侵袭性肿瘤中,库欣病的表现是温和的,甚至可能是静默的,其组织学表现为稀疏的颗粒性促肾上腺皮质激素细胞肿瘤。在最近的流行病学研究中,Mete等报道,180例促肾上腺皮质激素细胞肿瘤中有52例为稀疏颗粒性肿瘤,36例在临床上被诊断为无功能性的,10例与激素超量有关(6例的临床资料不完整)。他们是由轻度嗜碱性(lightly basophilic)或嫌色性(chromophobic)细胞组成,只显示稀疏性或没有PAS及ACTH阳性,而只检测到Tpit(图12)。电镜下肿瘤细胞呈多面体细胞,细胞器发育不良,分泌颗粒小而稀疏。与致密颗粒型不同,1型细丝在细胞质中非常少。
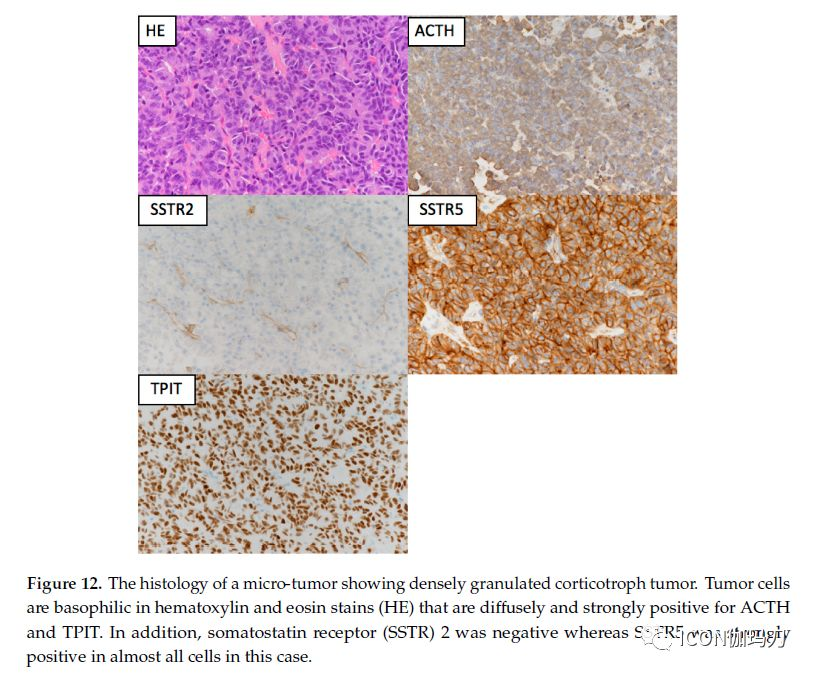
图12.微腺瘤的组织学表现为致密颗粒性促肾上腺皮质激素细胞肿瘤。肿瘤细胞的苏木精和伊红染色(HE)呈嗜碱性,对ACTH和TPIT呈广泛而强烈的阳性反应。此外,在本例中,生长抑素受体(SSTR) 2呈阴性,而SSTR5几乎在所有细胞中呈强阳性。
8.1.3.Crooke细胞肿瘤
Crooke玻璃样变(Crooke’s hyaline changes)一直被认为只在库欣病患者的正常垂体促肾上腺皮质激素细胞中发生,但最近的证据表明,在某些分泌促肾上腺皮质激素肿瘤的细胞中也观察到大量的玻璃样变。部分(超过50%的细胞)或全部由Crooke细胞组成的肿瘤称为Crooke细胞肿瘤[31]。这种类型的肿瘤通常表现出进袭性(aggressive)的临床病程,被认为是促肾上腺皮质激素细胞肿瘤的进袭性变异。电镜显示细胞质中充满了1型中间性细丝和分泌颗粒,它们被转移到质膜下区域(be displaced to the subplasmalemmal area)(图11)。虽然Crooke细胞肿瘤是一种罕见的亚型,在所有促肾上腺皮质激素细胞肿瘤中发病率估计为4.4-14%,但它们通常具有进袭性,表现为侵袭性大腺瘤,高复发率,对手术和放疗具有抵抗性。促肾上腺皮质激素垂体癌常发生于存在Crooke细胞肿瘤中。
8.2.进袭性垂体肿瘤和垂体癌
垂体癌被定义为有颅脑脊髓和/或全身转移的肿瘤。垂体癌的病理特征与典型或进袭性肿瘤的病理特征的区别并不明显,唯一的显著特征是存在转移。它们很罕见,占所有垂体肿瘤的0.1%-0.2%。大部分的垂体癌来源于促肾上腺皮质激素细胞瘤或泌乳素瘤[309]。先前的世界卫生组织(WHO)分类将垂体肿瘤分为典型肿瘤、不典型肿瘤和垂体癌。然而,在最近更新版的WHO分类中,因为不能提供组织病理学发现与临床行为之间的准确相关性,“不典型肿瘤”一词被删除,。根据临床表现,垂体瘤可分为进袭性和非进袭性。一些肿瘤表现出预测复发和对常规和多模式治疗的抵抗性的特征。这些特性包括迅速生长,放射影像学上的侵袭性,Ki-67标记指数高。这些肿瘤被称为临床进袭性肿瘤,必须进一步研究和密切随访。目前,尚无证据表明垂体癌的发生与特定的遗传或表观遗传背景或共同因素有关。最近的荟萃分析中,垂体癌与进袭性肿瘤相比,现实与很多因素相关:刺激细胞生长,血管生成,和侵袭性(细胞周期蛋白[cyclin]D1、VEGF[血管内皮生长因子、MMP[基质金属蛋白酶]9, miR-122,和miR-493)被认为是被调节的,而几个因素,包括生长抑制剂和细胞凋亡诱导物(p16Ink4A分子,p27Kip1、MT3 bcl - 2、Bax, Bcl-X,和MGMT)被下调。据推测,这些变化的累积,而不是导致垂体癌发生癌特异性突变或表观遗传改变,与垂体肿瘤侵袭性的增加有关。Trouillas等人提出了通过侵袭性和增殖进行分级的一种新的临床病理分类方法,并在一项回顾性多中心病例对照研究中证明其有效性。此外,最近,国际垂体病理学俱乐部提出了一个新的术语,垂体神经内分泌肿瘤,与用于其他神经内分泌肿瘤相一致,并且认识到这些肿瘤的高度变异对患者的影响,因为目前的垂体前叶的肿瘤分类不能准确反映行为的临床表现。
8.3.术后药物治疗库欣病的知识
研究SSTR2和SSTR5、多巴胺2受体的表达,或免疫组化检测的MGMT(甲基鸟嘌呤甲基转移酶),可以在需要选择更合适的药物辅助治疗时提供有用的信息。大多数糖皮质激素肿瘤(>85%)表达SSTR2和SSTR5 mRNA, SSTR1 mRNA表达较少(63%)(图12)。SSTR亚型的膜密度,尤其是SSTR2,受到皮质醇增多症的影响,而SSTR5的表达相对不受高皮质醇水平的影响。因此,SSTR5在活动性库欣病患者中比SSTR2表达更为明显。Takeshita等人认为,USP8突变的存在可能预示着对帕瑞肽(pasireotide)的良好反应,这与SSTR5显示出较高的关联性。在垂体肿瘤中,MGMT的表达而不是MGMT启动子的甲基化可以预测对替莫唑胺治疗的反应。非突变的进袭性肿瘤,如Crooke细胞瘤,可能对烷基化剂替莫唑胺反应较好,因为它们的MGMT表达明显较弱。
9. 结论
库欣病患者由于合并症、死亡率增加和健康相关生存质量(HRQoL)下降而承受了着很大的负担。最近有证据表明,在长期接触超量的糖皮质激素后,这些负担在库欣病“治愈”后仍然存在,至少有部分存在。因此,许多研究者强调快速诊断和治疗以及适当随访的至关重要性。然而,由于没有单一的检测方法可以准确诊断库欣病,因此诊断往往具有挑战性,需要根据每位患者的需求,结合适当的生物学和影像学检查,逐步进行。库欣病的治疗是内分泌学中最具挑战性的任务之一。在某些情况下,即使采用安排有序的阶梯式的诊断方法,也很难将库欣病从其他库欣综合征的潜在病因中鉴别出来。手术仍然是几乎所有的库欣病患者的首选治疗方法。大部分肿瘤为库欣病的微腺瘤,所以TSS(经蝶切除手术)是合适的治疗方法;然而,一些大腺瘤瘤需要扩大TSS(经蝶切除手术)或联合(经蝶切除手术)和TC(开颅手术)的方法,以安全地最大限度地切除肿瘤。但是,尽管使用了现代MRI方法,一些(约10%的病例)治疗在MRI上是不可见的肿瘤。手术失败后持续或复发的库欣病需要进一步的治疗,包括重复手术、药物治疗、放射治疗,少数患者还需要接受双侧肾上腺切除术。重复手术应该只考虑针对那些在MRI上可以看到明显的、可以切除的肿瘤的患者,但通常与相对较高的失败率和复发率相关。目前可用的药物治疗包括肾上腺类固醇生成抑制剂,针对肿瘤的药物和糖皮质激素受体拮抗剂。肾上腺类固醇生成抑制剂对短期控制皮质醇过多是有用的,但经常发生多种副反应和药物相互作用。针对垂体肿瘤(tumor-directed)的药物具有潜在的缩小肿瘤,以及改善皮质醇增多症的优势。联合治疗是另 一 种策略来用增加治疗的疗效,同时使副反应最小化。一些新的药物治疗正在临床试验中,还有一些新兴的药物治疗目前正在评估中。放射治疗是有效的二线治疗,尤其当存在手术不能切除的肿瘤时,在进行放射治疗时,必须经常考虑到较长时间的延迟起效和与放射相关的副作用。相反,双侧肾上腺切除术能提供永久性的皮质醇增多症的治愈,但慢性肾上腺皮质功能不足和发生Nelson综合征的风险一直备受关注。总的来说,一个专家协作的多学科的团队的管理这一复杂疾病的强制性要求,以个人为基础的方法是必不可少的,以达到最高的成功,同时尽量减少副作用和改善患者的生存质量。最后,是备受期待的,最近的在分子水平上对库欣病的病理生理学的新见解,将为在不久的将来,给这种可怕的疾病带来诊断检测的准确性和更为有效的治疗。
