


《Endocrinology and Metabolism Clinics North America》杂志 2020年9月[ Sep;49(3):433-452.]刊载英国Queen Mary University of London的Barry S 和 Korbonits M 撰写的综述《垂体瘤的遗传学进展。Update on the Genetics of Pituitary Tumors.》 (doi: 10.1016/j.ecl.2020.05.005.)。

垂体腺瘤是颅内常见的肿瘤,表型多样。这些肿瘤大多数是偶发的,不是遗传疾病的一部分。在过去的几十年里,大量的遗传学研究带来对与垂体肿瘤相关的体细胞和种系突变的识别,这提高了对垂体肿瘤发生的理解。探索垂体神经内分泌肿瘤的遗传背景可导致早期诊断和较好的预后,其分子机制应导致新的靶向治疗,即使是零星肿瘤。本文就垂体瘤的相关基因和综合征作一综述。
●垂体神经内分泌肿瘤(PitNETs)通常是颅内良性肿瘤,很少转移瘤。95%以上的肿瘤在起源时是散发性的,约5%含有种系突变引起综合征或孤立性垂体腺瘤。
●在过去的几十年里,大量的遗传学研究发现了与垂体肿瘤相关的体细胞突变(GNAS, USP8, GPR101)和种系突变(MEN1,细胞周期蛋白依赖性激酶抑制基因,AIP, DICER1, PRKAR1A, PRKACA, SDHx,和GPR101),这促进了对垂体肿瘤发生的理解。
●我们概述了垂体肿瘤的遗传学。
●目前的挑战和暗示这些基因发现在临床实践中被讨论。
在一般人群中,有症状的垂体神经内分泌肿瘤(PitNETs)的发病率约为千分之一。这些肿瘤大多数是偶发的,不属于综合征异常(syndromic disorders)的一部分。估计有5%的人被认为有家族性起源。少数有明确的遗传综合征已知与垂体腺瘤有关,包括多发性内分泌瘤(MEN)1型和Carney复合体(CNC),以及非综合征性家族性孤立垂体腺瘤(FIPA)。PITNETs生长缓慢,通常是良性肿瘤,一般认为是单克隆起源。然而,由于对周围组织的肿块占位效应或激素分泌的改变,它们可以引起相当多的并发症发生率。此外,肿瘤可侵入周围结构,导致完全手术切除的困难。对于某些肿瘤,药物治疗可用生长抑素类似物或多巴胺激动剂,而对于其他肿瘤,手术和放疗是唯一的治疗选择。了解PitNETs的遗传背景,可以带来早期诊断和更好的结果,其分子机制应该引导甚至对散发性肿瘤新的靶向治疗。我们在此总结与垂体瘤相关的基因和综合征。
尽管有大量的经典和最近的研究,散发性垂体腺瘤的特征是只有两个基因GNAS和USP8(泛素特异性肽酶8)反复发生的体细胞突变(图1)。在GNAS、GPR101和最近的MEN1中发现了一种独特的体细胞突变,即胚胎突变导致的嵌合体(embryonic mutations leading to mosaicism)。
该基因最常发生的体细胞杂合功能获得突变见于约40%的生长激素细胞肿瘤中。GNAS编码gsp癌基因,即刺激G蛋白亚基α (Gsα)。主要的突变发生在密码子201(codon 201),而密码子227发生突变,但远没有那么频繁。这种突变破坏Gsα的GTPase活性,导致腺苷环化酶的组成性激活(constitutive activation)和环磷酸腺苷(cAMP)水平的增加:cAMP结合蛋白激酶A的调节亚基(PKA;通过PRKAR1A/1B或PRKAR2A/2B编码,使催化亚基(PRKACA/B)增加PKA水平,进而激活cAMP反应元件结合蛋白(CREB)。CREB激活PIT1 (POU1F1)转录因子,促进生长激素(GH)高分泌,增加生长激素细胞增殖。GNAS是一个在垂体中的印记基因(imprinted gene),突变总是位于母系等位基因上。几项研究发现,gsp阳性的生长激素细胞肿瘤对生长抑素类似物治疗有较好的反应,具有明显的临床特征,包括诊断年龄较大、肿瘤体积较小、侵袭性性质较少以及组织学上为致密颗粒形态。 GNAS突变很少在其他肿瘤类型中被发现,在1 /21和1/14的无功能垂体腺瘤(NFPAs)和1例促肾上腺皮质激素细胞腺瘤中被发现。在生长激素细胞肿瘤中没有发现其他复发的体细胞突变。

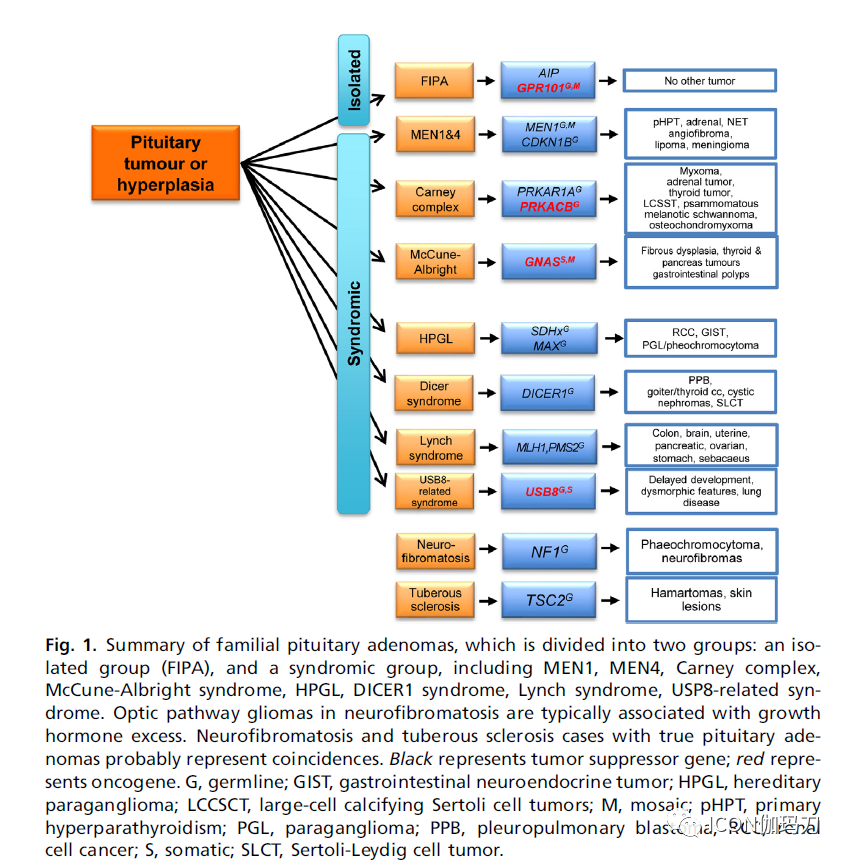
图1.家族性垂体腺瘤概述,分为两组:孤立性组(FIPA)和包括MEN1、MEN4、Carney complex、McCune-Albright综合征、HPGL、DICER1综合征、Lynch综合征、USP-8相关综合征综合征性组。神经纤维瘤病中视觉神经通路胶质瘤通常与生长激素过量有关。神经纤维瘤病和结节性硬化症患者合并真正的垂体腺瘤可能是巧合。黑色代表抑癌基因;红色代表致癌基因。G,生殖系;GIST,胃肠道神经内分泌瘤;HPGL,遗传性副神经节瘤;LCCSCT,大细胞钙化支持细胞肿瘤:mosaic,嵌合的;pHPT,原发性甲状旁腺功能亢进;PGL,副神经节瘤;PPB,脏层胸膜的胚细胞瘤;RCC,肾细胞癌;S,体细胞;SLCT,支持间质细胞细胞瘤。
如果GNAS突变发生在胚胎发育期间,就会发生McCune-Albright综合征(MAS)嵌合性疾病(见图1)伴有多样的表型,取决于哪些组织受到突变的影响。MAS是以多囊纤维发育不良(polycystic fibrous dysplasia)、性早熟(precocious puberty)和咖啡斑(cafe´ au lait spots)为特征的系统性疾病。这是由于GNAS基因在合子后期早期功能获得突变导致体细胞嵌合体。GNAS位于编码cAMP通路相关G蛋白Gsα的20q13染色体上,是在生长激素细胞腺瘤中发现的唯一复发性突变。与MAS相关的最常见的垂体肿瘤类型是生长激素细胞腺瘤(约30%),其他表现为垂体增生。MAS中最常见的GNAS突变是R201H或R201C。这些位点的突变导致GTPase活性的丧失,随之而来的是腺苷酸环化酶的永久激活和cAMP依赖的PKA通路的组成性激活。这导致包括垂体的cAMP反应组织的细胞增殖增加。患者的特征取决于受影响的组织。
最近发现,促肾上腺皮质激素腺瘤的USP8基因中存在周期性的体细胞获得功能突变(见图1)。该基因编码一种去泛素化酶,可抑制表皮生长因子受体(EGFR)的溶酶体降解,从而导致EGFR信号通路和细胞生长的增加。USP8的突变影响14-3-3蛋白结合结构域,该结构域通常保护USP8不被切割;突变阻断了14-3-3与被切割USP8的结合位点,显示出较高的泛素降解活性,从而导致更多的再循环的EGFR和通过这些受体增加的信号通路,然后上调POMC表达。然而,USP8突变状态并不总是与EGFR表达相关。最近一项对10项研究的677例患者的荟萃分析表明,32%的女性和24%的男性促肾上腺皮质激素腺瘤患者有体细胞USP8突变。尽管数据是多变的,这项荟萃分析发现USP8突变的促肾上腺皮质激素腺瘤的平均肿瘤大小为9毫米,其中42%的微腺瘤和31%的大腺瘤发生了突变。突变阳性的患者在术后有更好的保持缓解的机会。最近也有报道在一个16岁复发库欣病女孩杂合子种系USP8突变,包括发育迟缓,畸形特征,鱼鳞病样角化过度,慢性肺病和肾病,以及心肌病。
生殖系GPR101复制通常在女性可见,作为新生的突变(见图1)。到目前为止,所有的男性患者没有家族史的X连锁肢端巨人症(XLAG)已发现与嵌合的GPR101复制,而今天描述的三个有家族血缘的,发现男性后代伴有生殖系复制。到目前为止,还没有描述过女性伴有嵌合现象。种系与嵌合病例之间无表型差异。
最近报道了MEN1嵌合体突变的患者和种系MEN1的后代,强调在明显散发的MEN1新生突变患者中需要考虑嵌合体。
将与垂体腺瘤相关的种系突变患者分为两组:孤立性组和综合征性组(见图1)。
多发性内分泌瘤1型
MEN1是一种遗传性常染色体显性综合征,与垂体、甲状旁腺、胰腺肿瘤、面部血管纤维瘤、胶原瘤、肾上腺皮质腺瘤、脂肪瘤、脑膜瘤和罕见的嗜铬细胞瘤有关。在90%的患者中发现MEN1杂合钝化突变,并在11q13肿瘤中显示杂合性缺失,大约30% - 40%的MEN1患者发展为垂体瘤。泌乳素瘤是临床上最常见的垂体瘤(占临床诊断的MEN1垂体腺瘤的60%),其次是无功能垂体腺瘤(NFPAs)(15%-40%)、生长激素细胞垂体瘤(5%-10%),极少数是促肾上腺皮质激素细胞或促甲状腺激素细胞腺瘤。在MEN1患者中,垂体肿瘤被发现较大,并且在组织学上更具侵袭性,虽然这方面一直有争议。MEN1基因位于11号染色体长臂上,编码肿瘤抑制核蛋白menin。它在转录调节、基因组稳定性、细胞分裂和增殖过程中有许多相互作用的伙伴。到目前为止,已有超过1500种系突变得到描述,其中大多数是由无义突变和由于缺失或插入引起的移码突变所导致的截断突变。尽管有一些有争议的数据,没有明确的垂体肿瘤的基因型和表型之间的关系。
多发性内分泌瘤4型
MEN4是由CDKN1B或其他周期蛋白依赖的激酶抑制剂基因的种系杂合缺失功能突变引起的常染色体显性综合征(autosomal-dominant syndrome)。大约10%的MEN1样综合征患者没有MEN1突变,可能是由内含子或启动子的隐性突变或表型引起的。然而,一些可能显示出CDKN1B突变,从而代表一种MEN形态,命名为MEN4(多发性内分泌瘤4型)。垂体瘤发生在37%的报道的MEN4病例中。其他肿瘤包括甲状旁腺腺瘤、肾上腺瘤、肾血管平滑肌脂肪瘤、子宫平滑肌瘤、胃泌素瘤、神经内分泌子宫颈癌、支气管类癌、甲状腺乳头状癌和胃癌。CDKN1B位于染色体12q13且编码p27, p27是参与细胞周期调控的细胞周期依赖激酶抑制剂。CDKN1B突变包括移码突变、无义突变、错义突变和导致p27表达减少的50UTR突变。由于MEN4的罕见性,潜在的基因型表型的相关性仍有待证实。
Carney复合征(CNC)是一种罕见的常染色体显性综合征,其特征为皮肤色素沉着(skin pigmentation)、心脏黏液瘤(cardiac myxomas)、分泌生长激素和泌乳素的垂体腺瘤,以及由原发性色素沉着结节性肾上腺皮质疾病(primary pigmented nodular adrenocortical disease)引起的库欣综合征。最常见的Carney复合征(CNC)是由于编码PKA调节亚基1α型的位于染色体17的肿瘤抑制基因PRKAR1A基因的种系突变。突变灭活和PRKAR1A大量删除导致PKA 1α调节亚基功能损失,导致cAMP-依赖的 PKA活性增加。最常见的突变是无义突变和移码突变。在Carney复合征(CNC)患者中观察到基因型-表型的相关性(genotype-phenotype correlation),大量缺失会导致更严重的疾病。约75%的Carney复合征(CNC)患者显示GH轴异常;临床上有明显的肢端肥大症并不常见(10%-12%)。儿童期起病的生长激素(GH)过量导致巨人症的报道很少。泌乳素瘤是罕见的,而2例库欣病与Carney复合征(CNC)相关的病例已经得到描述。染色体2p16上的第二个位点也与Carney复合征(CNC)有关联,但是在这个位点还没有发现基因。PKA活动增加的结果不仅仅是调节缺失(即抑制活性、PRKAR突变的功能丧失),也因为激活催化亚基突变( 19号染色体上的PRKACA和染色体1上的PRKACB)。体细胞PRKACA点突变与分泌皮质醇的肾上腺腺瘤相关,而生殖系二倍体和三倍体(germline duplicationsand triplications)可以导致儿童期或成人起病的双侧的小结节或大结节增生。一例生殖系双倍体患者出现类似Carney复合征(CNC)的皮肤病变(内眦色素沉着[pigmented inner canthus],红唇边缘色素斑点[pigmented spots on the vermilion border],面部雀斑[facial freckling]),但无其他与Carney复合征的相关表现,而其他患者无皮肤表现。一例典型的Carney复合征报道有PRKACB重复。因为肾上腺皮质腺瘤和生长激素细胞腺瘤患者,依赖于CAMP信号通路,在散发性生长激素细胞腺瘤和其他类型的垂体腺瘤中测试热点突变体细胞L206R影响PRKACA和PRKACB;在垂体瘤组织中未识别出变化。
家族性孤立性垂体腺瘤(FIPA)是一种常染色体显性疾病,其定义为在同一家族的两个或两个以上家族成员中存在垂体腺瘤,且无其他综合征特征。到目前为止,在90%的家族性孤立性垂体腺瘤(FIPA)家族中,导致遗传易感性的基因还没有被发现。然而,在10%的FIPA家庭中已在芳烃受体相互作用蛋白(the aryl hydrocarbon receptor interacting protein,AIP)基因中识别出杂合子生殖系功能丧失突变(heterozygote germline loss-of-function mutations),而因为不完全外显率(incomplete penetrance),在明显的散发性病例、3.6%未经选择的零星的垂体腺瘤患者和,10%到20%的儿童垂体腺瘤患者中也发现了这种基因。大多数AIP突变阳性的患者临床表现为生长激素细胞或生长激素泌乳素细胞肿瘤,约10%为泌乳素细胞肿瘤,虽然免疫染色上生长激素或泌乳素通常呈阳性,很少出现临床无功能性腺瘤。有单例的促甲状腺素腺瘤(TSHoma)病例,而在有明确的AIP变异病变的促肾上腺皮质激素细胞腺瘤患者中没有被描述过。AIP突变阳性肿瘤与AIP阴性和散发性肿瘤具有明显的临床特征。在AIP突变阳性患者中,发病年龄较轻(平均诊断年龄为18-24岁),最小病例在4岁时诊断。如果在30岁时影像学和激素水平正常,我们还没有发现一例AIP突变阳性病例,该肿瘤的诊断年龄超过30岁。AIP突变阳性患者中,44%有巨人症。患者通常表现为大腺瘤(约90%)和常为侵袭性肿瘤(>50%)。约占所有病例中的8% 到10%的患者出现垂体卒中,其中以儿童期垂体性卒中为独特表现。散发性生长激素细胞腺瘤中AIP蛋白水平的降低与肿瘤的侵袭性有关。与散发性肿瘤相比,AIP突变阳性肿瘤显示免疫浸润数量增加。AIP突变患者需要多模式治疗,而不仅仅是一次手术。AIP突变阳性肿瘤对生长抑素类似物的治疗表现出相对的耐药抵抗性,并且在对生长抑素类似物耐药的散发性肢端肥大症患者中发现AIP突变的发生率增加。对高风险亲属(at-risk relatives)的遗传学筛查可以识别未受影响的携带者,他们将受益于临床筛查,这可能导致明显无症状的受试者得到早期诊断,引发必要时的早期干预。前瞻性诊断的突变携带者应按照现行指南管理。前瞻性诊断为AIP突变阳性的患者,与有临床表现的患者相比,病变较小,海绵状窦侵袭较少,较少需要多模式治疗。在前瞻性家庭筛查中识别的肿瘤谱与临床表现的病例不同:它们显示45%有GH超量;12%为泌乳素瘤,多为微腺瘤,无侵袭性;41%为临床无功能微腺瘤。这项由Marques和同事们所进行的研究,包括12年的随访,强调了基因和临床筛查的潜在获益。
AIP位于染色体11q13.2上,包含6个外显子,编码330氨基酸蛋白。目前已鉴定出超过100个AIP变异,其中包括缺失、插入、片段重复、无义、错义、剪接位点和启动子突变,以及整个外显子或整个AIP基因的大量缺失。突变存在于整个基因长度并破坏蛋白质。已知的AIP变异中有70%会导致蛋白质的截短(truncated)。大多数错义突变通常聚集在蛋白质的C末端,被认为是其生物功能的关键。临床和功能数据支持其作为肿瘤抑制基因的作用。在AIP突变阳性肿瘤中发现杂合性缺失。
垂体腺瘤在AIP突变携带者中的低外显率,以及其异质性的临床表型,提示有其他疾病修饰基因的参与。
到目前为止,在散发的垂体腺瘤中还没有发现AIP的体细胞突变。AIP低表达的散发性垂体肿瘤侵袭性增强。其他肿瘤如结直肠癌、乳腺癌、前列腺癌、内分泌肿瘤(甲状腺病变、肾上腺病变、类癌、甲状旁腺病变、副神经节瘤、胰腺内分泌肿瘤、腺癌)中,未发现AIP的体细胞突变。
AIP是一种分子协调伴侣蛋白(molecular cochaperone protein),与大量的伙伴(partners)相互作用,可能在许多重要通路中发挥作用。虽然杂合的功能缺失突变与生命完全相容,而且只有大约20%的携带者发生肿瘤,通常在一种特定的细胞类型(生长激素细胞)中,纯合敲除动物模型(小鼠、新杆状线虫Caenorhabditis elegans和果蝇Drosophila)是致死性的,表明其在生理上的关键作用。
第一个确定的AIP伙伴是核的异型生物受体AhR(芳基烃受体)。AIP调节其亚细胞定位和降解。其他互动的伙伴包括其他核受体(AhR, GR、PPARα,ERα,和TRb1),病毒蛋白(HBV-X和EBNA-3), G蛋白(Gα13和Gαq),分子伴侣蛋白质(Hsp90和Hsp70),磷酸二酯酶(PDE4A5、PDE2A3 和PDE3 / SLFN12),线粒体蛋白质(TOMM20),细胞骨架蛋白(Actin,TUBB和TUBB2A),和其他(RET,survivin,TNNI3K、TIF-2和 p23)已得到确定,这可能表明,AIP参与多种多样的细胞通路;然而,这些相互作用的后果还没有被完全理解。
根据实验发现,AIP参与CAMP依赖的PKA通路,而PKA通路在调控生长激素的表达和体细胞增殖中发挥重要作用。AIP与调节cAMP通路的磷酸二酯酶4A4/5相互作用,cAMP通路的激活在体细胞肿瘤发生中是重要的。AIP最近还显示与参与细胞骨架组织的蛋白质相互作用。
AIP纯合缺失对胚胎是致命的,因为心血管发育异常和红细胞生成失败,提示AIP在心脏发育和维持小鼠红细胞生成中发挥关键作用。AIP基因的杂合子缺失导致生长激素和泌乳素分泌垂体腺瘤的发生。AIP缺陷小鼠模型概括了人类的表型。另一种小鼠模型已经建立,AIP只在体细胞中被删除。在12周龄时的AIP敲除小鼠比野生型对照组大,在18周龄时小鼠的GH和IGF-1水平显著升高。在20周龄的敲除小鼠中,80%的小鼠可以看到肉眼可见的肿瘤。
最近,X连锁肢端肥大巨人症(XLAG)被描述为有生长激素超量的早期发病的患者的罕见的情况。通常在X染色体上的一个复制区域(Xq23.6)内,GRP101基因的新种系复制或嵌合复制(De novo germline or mosaic duplication)。虽然最初识别的Xq26.3重复区域涉及4个基因,但在垂体组织中发现只有GPR101基因在mRNA水平上调,且在1例患者中仅该区域被重复,使得该基因在表型中极有可能发挥致病作用。GPR101基因编码G蛋白偶联受;虽然之前发现了一种孤儿受体,最近发现了一种内源性配体,脂质分子Resolvin D5, 但该配体在GH调节或XLAG发病机制中的作用尚不清楚。XLAG综合征的特点是超量GH生产和早期发病的加速增长,在大多数患者中,观察到在生命的头两年,在所有的病人在4岁之前,常伴有肢端肥大症的症象(粗陋的面部特征coarse facial features、出汗、头痛)伴食欲增加/求食行为(food seeking behavior.)。XLAG相关垂体肿瘤表现为典型的窦状和小叶状结构(sinusoidal and lobular architecture),常伴有钙化和滤泡样结构。因为对治疗具有抵抗性,疾病控制是具有挑战性的,往往需要完全切除垂体,或复杂的药物治疗,以及多巴胺激动剂、生长抑素类似物、生长激素受体拮抗剂,和放射治疗。
在巨人症患者中,XLAG的患病率为4.4%到10%。多为女性。在这些分泌GH的垂体肿瘤中,伴有鞍上伸展和海绵窦侵袭的大腺瘤明显更为常见。垂体增生约占25%。高达85%的患者可出现高泌乳素血症。两项对巨人症患者的大型研究共确认了31例由XLAG引起的垂体腺瘤或垂体增生。迄今报告不到40例。在三个家庭中,已观察到母亲传给儿子的全外显(full penetrance)是引起家族性孤立性垂体腺瘤(FIPA)的一种罕见原因。GPR101在XLAG患者的垂体肿瘤中过表达,但在散发性生长激素细胞肿瘤或成人垂体腺中不表达。尚不清楚XLAG的潜在发病机制。在先天性孤立性生长激素缺乏症患者中未发现致病性GPR101突变或拷贝数变异。
DICER1是一种罕见的常染色体显性遗传病,由DICER1基因种系杂合突变引起。DICER1的突变导致多种肿瘤的表现,包括脏层胸膜的母细胞瘤(pleuropulmonary blastoma)、卵巢性素间质细胞肿瘤(ovarian sex cord-stromal tumors)(主要是Sertoli-Leydig细胞肿瘤),囊性肾瘤(cystic nephroma),甲状腺结节性增生,分化型甲状腺癌,垂体母细胞瘤、鼻软骨间叶性错构瘤(nasal chondromesenchymal hamartoma),睫状体髓质上皮瘤(ciliary body medulloepithelioma),肾肉瘤,泌尿生殖胚胎性横纹肌肉瘤(genitourinary embryonal rhabdomyosarcoma),和松果体母细胞瘤(pinealoblastoma)。垂体母细胞瘤是低外显率(1%)的DICER1综合征的罕见表现。他们在婴儿期(通常小于2岁,伴分泌促肾上腺皮质激素的垂体母细胞瘤)由DICER1基因突变引起。之所以称为“母细胞瘤(blastoma)”,是因为这些个体的垂体腺看起来类似于胚胎组织。DICER1位于染色体14q32.13上:这是一种RNA裂解酶,可以将前体microRNA裂解为成熟的miRNA;miRNAs是控制特定RNA分子表达和/或降解的调控蛋白。
DICER1突变患者的肿瘤经常显示有生殖系功能丧失的等位基因的突变,在另一个等位基因中的第二个打击性致病性体细胞变异(hit pathogenic somatic variant)通常在DICER1的催化RNaseIII a和IIIb结构域的金属结合位点内。嵌合RNaseIII a和IIIb结构域突变与严重的表型相关,但至今在这些患者中没有垂体母细胞瘤的描述。最近,报道1例泌乳素微腺瘤患者的种系DICER1突变;在这一点上,这看起来更像是一种巧合,而不是两种疾病之间有因果关系(causative relationship)。
家族性副神经节瘤(familial paragangliomas)、嗜铬细胞瘤(pheochromocytomas)和垂体腺瘤(pituitary adenomas)的关联,被称为“3P关联(the three P association)”,是种系SDHx突变引起的罕见的情况。在SDHx中垂体腺瘤的患病率很低,约为0.3%;然而,这可能不是真实的表现,因为SDHx突变携带者没有对垂体肿瘤作常规检查。所有琥珀酸脱氢酶(the succinate dehydrogenase, SDH)基因突变的患者中已报道存在垂体腺瘤。SDH相关垂体腺瘤的一个独特特征是免疫组化分析显示胞质内空泡(the intracytoplasmic vacuoles),这与自噬体(autophagic bodies)的存在相对应。与SDH相关的垂体肿瘤主要是分泌PRL或分泌GH或是无功能性腺瘤,而且更可能对标准化治疗产生耐药抵抗性,表现得更具有进袭性,还有一例转移性病例也得到描述。尚不完全清楚失去SDH功能导致肿瘤发生的确切机制。
SDH蛋白复合物由多个亚基(SDHA、SDHB、SDHC和SDHD)及其相关组装因子(assembly factor)(SDHAF2)组成。SDH与线粒体内膜结合,是Krebs循环的一员,参与氧化磷酸化(oxidative phosphorylation)。这些亚基的任何基因突变都可能导致电子传递链受损,从而导致代谢物的积累和缺氧诱导因子-1a的最终积累,从而导致血管内皮生长因子上调。随后,缺氧诱导因子-1a负责引起对凋亡信号的抵抗,并增强肿瘤中的糖酵解。此外,垂体腺瘤和副神经节瘤/嗜铬细胞瘤的关联也出现在其他遗传综合征中,如与MEN1、RET、VHL和MAX基因突变有关的综合征。
CDH23基因的杂合错义突变已在一个家族性孤立性垂体腺瘤(FIPA)亲属(kindred)中被发现,其中有两例肢端肥大症和两例无功能性垂体腺瘤(NFPA)。 CDH23编码与Cadherin相关的23,它是cadherin超家族的成员,参与细胞间的黏附。在125例散发的垂体肿瘤中发现了15例罕见的CDH23变异,并预测其可能致病。纯合CDH23突变与耳聋和Usher综合征相关。然而,没有功能性数据可以解释这些突变是如何参与垂体肿瘤发生的,也没有垂体腺瘤被描述与Usher综合征有关,因此尚不清楚它们对垂体肿瘤发生的致病性。
在182例名库欣病患者的队列中,4名散发性促肾上腺皮质激素细胞腺瘤患者(2例年青的成人和2例小儿)中发现有种系CABLES1 (CDK5和ABL1酶底物1)变异。然而,没有发现家族性疾病。CABLES1位于染色体18q11.2上,编码一种蛋白质,该蛋白质是糖皮质激素作用下促肾上腺皮质激素细胞激活的细胞周期进程的负调控因子。CABLES1蛋白在约50%的促肾上腺皮质激素细胞腺瘤中缺失。需要进一步的验证性研究来评估库欣病或其他垂体肿瘤患者中CABLES1突变的发生。
神经纤维瘤病1型是一种罕见的常染色体显性多系统遗传疾病,由NF1基因失活突变引起,表现为良恶性肿瘤。这与皮肤神经纤维瘤,咖啡牛奶皮肤病变,和檫烂性雀斑(intertriginous freckling.)有关。约10%的神经纤维瘤1型和视神经胶质瘤患者临床表现为肢端肥大,GH和IGF1超量的特点,但无可见垂体病变。NF1编码神经纤维蛋白;作为Ras - GTPase(酶)激活蛋白;NF1的缺失导致特别是Ras/Raf/MEK和Ras/ PI3K/TSC/mTOR等Ras依赖通路的组成性激活。有报道称,一位伴有分泌GH腺瘤的NF1患者没有杂合性缺失、NF1基因位点和GNAS突变,提示NF1在垂体肿瘤发生中没有作用。需要进一步的研究来确定NF1突变和垂体肿瘤之间的联系。
结节性硬化症是由TSC1和TSC2基因的遗传缺陷引起的常染色体显性综合征,这两个基因分别编码hamartin(错构瘤蛋白)和tuberin(马铃薯球蛋白)。TSC1/ TSC2介导PI3K/Akt的激活并导致mTOR通路的抑制。结节性硬化症表现为错构瘤、癫痫和智力迟钝(mental retardation)。到目前为止,只发现4例可能有突变的TSC和垂体肿瘤的患者,2例是促肾上腺皮质激素,1例是生长激素,和1例静默性促性腺激素肿瘤。同样,这些情况也可能是巧合。
与其他基因筛查指南(如乳房和结肠)类似,基因筛查指南对患有垂体神经内分泌肿瘤(PitNETs)的患者的应用具有挑战性。需要考虑以下三个问题:(1)哪些PitNET患者应该筛查其中一个垂体肿瘤易感基因;(2)对携带者的家庭成员应该做什么临床筛查;和(3)确定致病性/可能致病、或良性/可能良性的测序改变的变异的分类。
(一)散发性垂体腺瘤病例中种系突变的频率较低,因此不建议对所有患者进行种系变异的基因筛查。当有有提示存在一种综合征,然后适当的基因或基因固定样本(panel)可以用于筛查。一个直接的情况是MEN1患者或有类似MEN1的临床表现,并有阳性的家族史或存在该综合征的其他表现的患者。由于MEN1可能是新生突变,而垂体腺瘤可能是该疾病的第一个表现,因此对仅有垂体神经内分泌肿瘤(PitNET)的患者进行检测时需要仔细判断。在我们看来,对进袭性生长的儿童期泌乳素瘤患者检测MEN1突变证明是合理的。
Crney复合征(CNC)通常是一种基于渊源者(proband)表现的临床诊断。外显率为100%,通常根据成年人年龄:即使在家族环境中,新生突变也得到很好地描述。在特殊的SDHx阳性患者中,垂体也可能是唯一的表现。在非综合征性疾病中,GPR101通常可通过临床特征识别(如年轻起病的垂体巨人症[年龄<5岁]),对这些患者应进行GPR101重复基因检测(genetic testing for GPR101 duplications)。即使是小的重复,应该谨慎地使用技术来识别种系突变,或对垂体或其他组织的嵌合突变进行测试。
对于散发性或家族性年轻起病的生长激素超量,AIP筛查是关键的,但由于10%的AIP阳性病例是泌乳素瘤,我们也建议对年轻起病的泌乳素大腺瘤进行检测(虽然泌乳素微腺瘤也有伴有AIP无义突变的报道)。起病年龄较大(>30岁)的家族性病例不太可能是阳性的,但目前仍处于与AIP相关研究的发现阶段,我们建议筛查所有家族性孤立性垂体腺瘤(FIPA)患者,至少是最年轻的受影响的家庭成员。人们需要意识到在AIP家族中描述的 拟表型(phenocopy,一种环境影响引起的表现型非遗传性变更)。基因检测正在向固定样本(panel)检测发展,目前有几个实验室提供了这种选择,包括大多数垂体肿瘤相关基因。
对识别出突变阳性的家庭成员进行初步临床筛查很重要,因为在这个阶段就会发现已经存在的异常,这显然取决于具体的疾病和患者的年龄。对明显未受影响的携带者家庭成员的进一步随访具有挑战性。尽管密集的临床随访可以在早期发现疾病,但它有诱发筛查疲劳的潜在危险,患者和家人可能在疾病发展风险变得很高的时候脱离筛查。终生监测在时间、频率、成本、偶然发现和携带者的焦虑等方面具有重大意义。对综合征疾病基因携带者的全面随访的讨论超出了本综述的限制。
对于MEN1综合征的垂体方面,建议从5岁开始筛查,每年进行垂体相关激素的血液检查,并每三年进行一次磁共振检查。对于Carnry复合征(CNC)的垂体方面,由于垂体相关的异常主要发生在GH/PRL轴,根据垂体功能和基线垂体MRI,每年进行GH、PRL和IGF-1的血液检查,并建议进行临床随访。对于SDHx,突变阳性患者垂体疾病外显率低;常规的身体MRI筛查可在首次筛查时扩展到垂体窝,然后每3 - 5年一次。
由于DICER1突变阳性患者仅在2岁以下出现垂体异常,因此无需进一步垂体随访。对于AIP突变携带者,我们建议从4岁到童年和青春期仔细随访生长和青春期发育,除非需要更早,否则第一次MRI检查应在10岁左右进行。因为30岁后疾病不太可能发展,所以在这个年龄之后可以减少或停止筛查,尽管对这个年龄组的数据收集仍在进行中。由于外显率为100%,因此携带者随访不成问题。
对于AIP突变阴性的家族性孤立性垂体腺瘤(FIPA)的家族成员,由于疾病起病通常为中年,外显率低,且无法在临床研究之外识别携带者家族成员,因此没有特别的随访建议,尽管就可能的症状对患者/家庭成员进行教育是很重要的。
(二)对已识别的变异进行分类是临床遗传学家面临的最大挑战。许多在电脑模拟的(silico)预测程序现在是可用的和从这些(例如,www.varsome.com)在线资源结合数据是有帮助的。分类变量根据美国医学遗传学和基因组学学院对致病的、可能致病的,不确定意义的变异、可能良性和良性的指南(the American College of Medical Genetics and Genomics guidelines to pathogenic, likely pathogenic, variant of uncertain significance, likely benign, and benign)用于制定决策,但是需要明白这些数据可能会改变和持续的警惕和准备需要改变类别,特别是对于不确定意义的变化的大群变异(the large group of variant of uncertain significance changes)。要把这种复杂且往往不确定的信息传达给病人和他们的家人是很有挑战性的。
