神外资讯【中华神外】专栏,每周发布一篇精选文章,今天刊登的是由解放军总医院第一医学中心神经外科攸娜、刘羽阳、张军、许百男在《中华神经外科杂志》2019年第九期“病例报告”上发表的“施万细胞瘤病的基因诊断一例并文献复习”,欢迎阅读。
摘要
施万细胞瘤病是神经纤维瘤病的第3种亚型,由于肿瘤多发且易复发,临床特征不明显,极易被误诊为2型神经纤维瘤病等疾病,给临床诊断和后续治疗带来挑战。研究发现,SMARCB1和LZTR1基因与其发病密切相关。本文报道了1例临床表现为全身多发包块、颅内有占位性病变的神经纤维瘤病患者,对其行颅内占位性病变切除术,术后病理学诊断为神经鞘瘤,取外周血行基因检测,结果显示LZTR1强阳性,符合施万细胞瘤病的诊断特点。
施万细胞瘤病又称3型神经纤维瘤病(neurofibromatosis type 3,NF3),是一种常染色体不完全显性遗传性疾病[1],临床较罕见,年发病率约0.58/1000000[2],无明显性别差异,多成年发病,特征为多发性非皮肤侵犯的神经鞘瘤,同时不伴双侧前庭神经鞘瘤[3-4]。研究发现,施万细胞瘤病的发病与抑癌基因SMARCB1和LZTR1的突变有关[5]。由于其缺乏特异性的临床表现,在临床上极易被漏诊、误诊。本文报道1例施万细胞瘤病,并结合文献对该病的临床特点、诊断与鉴别诊断、治疗等方面进行探讨。
病历资料
患者 女,62岁,主因“体检发现颅内占位性病变2个月余”于2018年11月入住解放军总医院第一医学中心神经外科。患者30年前行左乳皮下包块切除术、2018年行左颈部包块切除术,病理学检查结果均为神经鞘瘤。入院后行正电子发射计算机断层显像(PET)-CT全身扫描,可见左颈部、盆腔左侧髂腰肌外侧、左侧臀部和双侧下肢近段的软组织及延髓水平椎管内多发高代谢灶,考虑为神经源性肿瘤;行头颅MRI检查,可见延髓右侧1个大小约1.3cm×2.1cm的类椭圆形病变,延髓稍受压,T1加权成像(T1WI)和T2加权成像(T2WI)均呈等或稍长信号,弥散加权成像(DWI)呈等信号,增强扫描后明显呈不均匀强化(图1A~D)。体格检查可见,左胫前部1个大小约2cm×2cm的皮下包块,明显隆起,与周围边界清晰,触之疼痛(图1E);右臀部1个大小约1cm×1cm的皮下包块,边界清晰,触诊时皮肤可在肿块上自由滑动。结合患者既往病史和影像学资料,术前诊断为右侧枕骨大孔区占位性病变,神经鞘瘤可能性大;首先考虑为施万细胞瘤病,但其他神经纤维瘤病暂不能排除。手术采取俯卧位,经后正中入路,术中可见肿瘤位于小脑扁桃体下方,副神经脊髓根位于肿瘤外侧,探查可见其中1支为肿瘤起源,给予切断并行肿瘤完全切除。术后行头颅CT检查,可见术区无出血,病变全切除(图1F)。术后病理学检查为右侧枕骨大孔区神经鞘瘤(图1G)。进一步取患者外周血进行基因学检测,结果显示NF2阴性,SMARCB1阴性,LZTR1强阳性,符合施万细胞瘤病的基因学特点,确诊为施万细胞瘤病(图1I)。术后3个月复查头颅MRI(图1H),结果显示肿瘤完全切除,未复发。
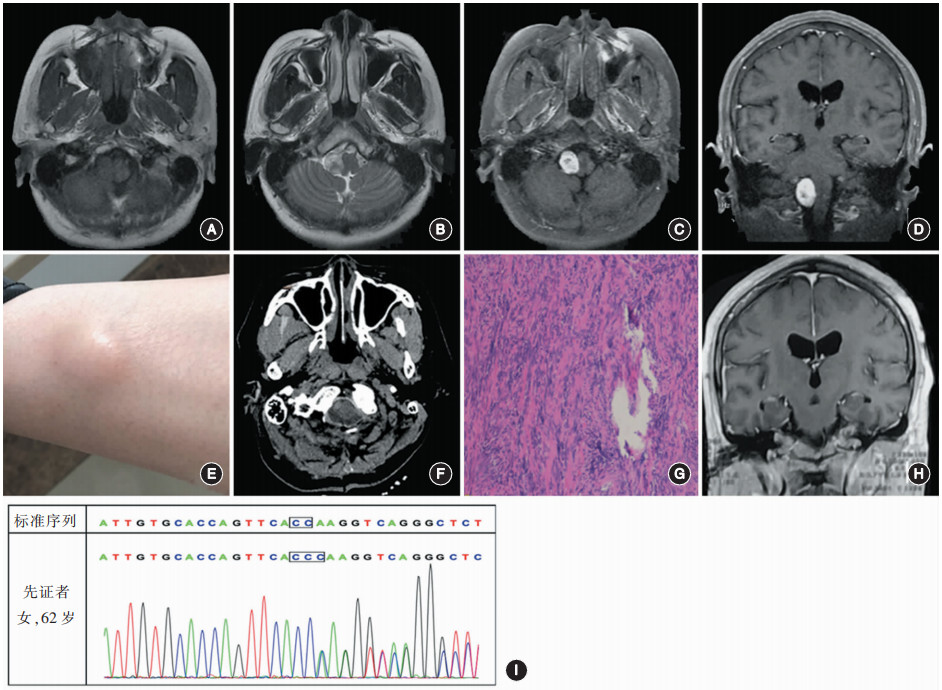
图1. 患者的影像学、病理学及基因检测资料;A,B.术前行头颅MRI检査,可见延髓右侧1个大小约1.3cmx2.1cm的类椭圆形病变,延髓稍受压,T1加权成像(T1WI)和T2加权成像(T2WI)均可见长信号;C,D.术前行头颅增强MRI检査,可见病变呈不均匀强化;E.体格检査可见,左侧小腿胫前存在边界清晰的皮下包块,触诊时皮肤可自由滑动,伴疼痛;F.术后次日行头颅CT检査,术区无出血,病变全切除;G.术后病理学检査为神经鞘瘤(HE染色xlOO);H.术后3个月复査头颅MRI,结果显示肿瘤完全切除,未复发;I.取患者外周血行基因检测,结果显示LZTR1基因编码区的2401和2402位之间插入1位碱基C,说明LZTR1基因发生了突变。
讨论
施万细胞瘤病是一种临床罕见疾病,特征为全身多发性神经鞘瘤,常累及周围神经和脊神经,脑神经较少受累。最常见为散发性,约20%的患者为家族性。本例患者既往2个手术史,病理学检查均为神经鞘瘤,但不满足2型神经纤维瘤病(neurofibromatosis type 2,NF2)的诊断标准,根据施万细胞瘤病的诊断标准确诊为施万细胞瘤病。但据此很难与嵌合型NF2进行区分,且制定早于LZTR1基因突变的发现,仍需进一步完善[6]。因此,对患者进行基因检测对于精确诊断施万细胞瘤病具有重要的指导性意义。
既往研究表明,SMARCB1和LZTR1基因的突变与该病密切相关[7-8]。SMARCB1基因位于22q11.23,编码具有抑癌作用的SMIRBC1蛋白,通过调节细胞周期和诱导衰老发挥肿瘤的抑制作用[9]。Sestini等[10]发现在具有SMARCB1突变的施万细胞瘤病患者存在“4次打击3次进展”的肿瘤发生模式。而不存在SMARCB1突变的施万细胞瘤病患者,常存在LZTR1突变。LZTR1基因也位于22q11.21,与SMARCB1(22q11.23)和NF2(22q12.2)相比更接近着丝粒。类似于SMARCB1基因突变的施万细胞瘤病患者,LZTR1基因突变的患者也存在类似的“4次打击3次进展”的肿瘤发生模式[11]。由于这两个基因位点均与NF2基因距离较近,因此大片段的缺失均可导致突变累及NF2,进而使患者出现类似NF2的病变。
本例患者因体检时发现颅内占位性病变入院进行手术治疗,术后病理学检查确诊为施万细胞瘤病。分析该例患者,30年前左乳皮下即出现包块,症状早于确诊时间,说明该病具有早期症状不明显、常出现延迟诊断的特点。而NF2患者早期便可出现单侧或双侧听力下降、伴或不伴耳鸣,症状较典型。施万细胞瘤病与NF2具有相似的影像学表现,因此难以通过影像学表现进行区分。但NF2患者以双侧前庭神经鞘瘤为特征,施万细胞瘤病则一般累及Ⅷ以外的脑神经,本例患者术中证实为副神经起源。施万细胞瘤病患者的全身多发性包块位于皮下,触诊时皮肤可自由滑动,伴疼痛,有别于NF2累及皮肤的固定性无痛包块。对本例患者取外周血进行基因检测,结果显示NF2阴性,SMARCB1阴性,LZTR1强阳性,符合施万细胞瘤病的基因学特点。神经纤维瘤病的治疗建立在精确诊断的基础上,3种亚型的病程和严重程度区别较大。与其他两型比较,施万细胞瘤病有一个相对良性的病程,治疗不宜激进,主要以对症治疗为主。关于施万细胞瘤病疼痛的治疗方法,应由经验丰富的多学科团队处理。若肿块进行性增大,产生较严重的压迫症状时可手术切除,但手术目的仅在于防止症状加重,对神经功能的恢复意义不大。放疗效果有限,常用于不适宜手术且有增大趋势的神经鞘瘤,但有诱发肿瘤发生恶变的风险。目前,化疗疗效尚不明确,不推荐使用。
综上所述,施万细胞瘤病与NF2的鉴别点如下:(1)发病年龄:施万细胞瘤病多见于中年,症状的出现时间远早于就诊时间,而NF2患者多于20岁左右。(2)受累神经:双侧前庭神经鞘瘤是NF2特征性的临床表现,而很少见于施万细胞瘤病。(3)疼痛特点:施万细胞瘤病患者常出现慢性疼痛,且可能与特定肿瘤无关,NF2无此特点。(4)基因检测:施万细胞瘤病患者常有SMARCB1或LZTR1基因突变,无NF2基因突变或合并NF2继发性突变。基于以上几点,可初步鉴别施万细胞瘤病与NF2。
目前,对于施万细胞瘤病的研究尚不全面,即使通过基因检测,也很难与不表现为前庭神经鞘瘤的嵌合型NF2进行鉴别。希望随着技术的进步及相关研究的发展,推进施万细胞瘤病规范化诊疗路径,结合基础研究,在未来该病的诊断与治疗可以达到新的水平。
参考文献