



神外资讯【中华神外】专栏,每周二发布一篇精选文章,今天刊登的是,由北京市神经外科研究所张亚卓所长、李储忠教授在《中华神经外科杂志》2018年第一期“标准与规范”上发表的《2017 版WHO垂体肿瘤分类解读》,欢迎阅读。
时隔13年之后,第四版《WHO内分泌肿瘤分类》于2017年6月出版,垂体肿瘤分类终于迎来了更新[1]。从临床角度看,新版最大的变化包括:
(1)将原来根据肿瘤细胞分泌激素的不同对垂体腺瘤进行分类转变为根据腺垂体细胞谱系对其进行分类,突出了调控垂体细胞分化的谱系特异性转录因子的作用(表1);
(2)取消了“非典型垂体腺瘤”(atypical pituitary adenoma)这一争议比较大的分类;
(3)对垂体非神经内分泌肿瘤进行了更多描述,引进了很多新分类。本文将主要针对新版的以上3点变化解读2017版WHO垂体肿瘤分类。
表1. 腺垂体细胞谱系及相关转录调控因子
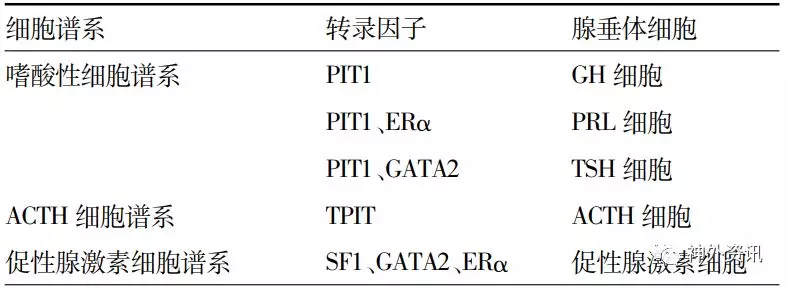
注:PIT1为垂体特异转录因子1(pituitary specific transcription factor 1);ERα为雌激素受体α(estrogen receptor α);GATA2为锌指转录调控蛋白GATA家族2;TPIT为T-box转录因子19;SF1为类固醇生成因子1(steroidogenic factor 1);GH为生长激素;PRL为催乳素;TSH为促甲状腺激素;ACTH为促肾上腺皮质激素
一、垂体神经内分泌肿瘤分类
(一)垂体腺瘤分类的新变化
前版WHO垂体腺瘤分类的基础是Kalman Kovacs发展的基于肿瘤细胞分泌激素类型不同进行分类的方法,结合患者的临床表现、分泌激素的类型和组织学表现3个方面综合考虑,并在临床广泛使用。该分类对临床无功能垂体腺瘤分类存在不足,尤其对于裸细胞腺瘤,如垂体激素免疫组织化学染色阴性就可以诊断,但部分裸细胞腺瘤有局灶α亚单位或其他激素的弱表达,通过相关转录因子染色发现大部分激素染色阴性的腺瘤来源于已分化的腺垂体细胞,如Nishioka等[2]认为,有95%的激素染色阴性的肿瘤表达腺垂体细胞谱系特异性转录因子,其中67%表达类固醇生成因子1(steroidogenic factor 1,SF1)和雌激素受体α(estrogen receptor α,ERα),提示该部分肿瘤来源于促性腺激素细胞谱系。
基于前版分类的缺点,新版改变了主要根据肿瘤细胞分泌激素的不同对垂体腺瘤进行分类的方法,转变为利用腺垂体细胞谱系特异性转录因子表达的不同确认肿瘤细胞的可能来源,结合激素表达情况进行分类并将其重新命名(表2)。例如将原有的生长激素(GH)腺瘤重新命名为GH细胞腺瘤,免疫组织化学染色除GH染色阳性外,还要有垂体特异转录因子(pituitary specific transcription factor 1,PIT1)的表达。对于不能确定肿瘤细胞起源的裸细胞腺瘤,除了垂体激素表达阴性,还要求各种相关转录因子表达阴性,使裸细胞腺瘤的诊断更为准确。
新版分类的另外一个特点是主要依赖免疫组织化学染色分类,首先通过垂体激素和转录因子表达确认肿瘤细胞的来源,然后应用
低分子量角蛋白(low molecular weight cytokeratin,LMWCK)染色的不同形式对肿瘤进一步分类,弱化了电镜超微结构在分类中的作用。新版分类指出,超微结构分析仅用于某些特定肿瘤的鉴别诊断,如PIT1阳性的多激素细胞腺瘤等。但笔者认为,在临床工作中,某些抗体免疫组织化学染色的可重复性不够好,尤其是那些尚未大规模临床应用的转录因子抗体,在这些情况下,结合超微结构进行鉴别诊断仍有较大价值。
垂体腺瘤临床功能分型及预后评估的相关因素在新版中也有小幅更新,增加了生长抑素受体2(somatostatin receptor 2,SSTR2)、SSTR5和O6-甲基鸟嘌呤-DNA-甲基转移酶(O6-Methylguanine-DNA methyltransferase,MGMT)以及错配修复基因6(mutS homolog6,MSH6)等预后预测指标(表3)。
(二)侵袭性垂体腺瘤(invasive pituitary adenoma,IPA)和难治性垂体腺瘤(clinical aggressive pituitary adenoma,CAPA)
表2. 2017版WHO垂体腺瘤的分类

注:GH为生长激素;PRL为催乳素;ACTH为促肾上腺皮质激素;LMWCK为低分子量角蛋白(low molecular weight cytokeratin);TSH为促甲状腺激素;FSH为卵泡刺激素;LH为黄体生成素;PIT1为垂体特异转录因子1(pituitary specific transcription factor 1);ERα为雌激素受体α(estrogen receptor α);GATA2为锌指转录调控蛋白GATA家族2;TPIT为T-box转录因子19;SF1为类固醇生成因子1(steroidogenic factor 1)
表3. 垂体腺瘤临床功能分型及预后评估的相关因素
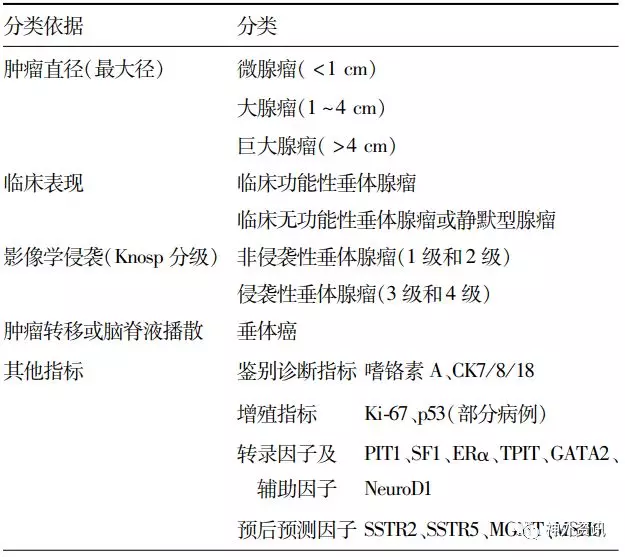
注:CK为细胞角蛋白(cytokeratin);PIT1为垂体特异转录因子1(pituitary specific transcription factor 1);SF1为类固醇生成因子1(steroidogenic factor 1);ERα为雌激素受体α(estrogen receptor α);TPIT为T-box转录因子19;GATA2为锌指转录调控蛋白GATA家族2;NeuroD1为神经分化因子1;SSTR2、SSTR5为生长抑素受体2和5;MGMT为O6-甲基鸟嘌呤-DNA-甲基转移酶;MSH6为错配修复基因
虽然垂体腺瘤为良性肿瘤,但仍有部分肿瘤破坏硬膜、骨膜甚至骨质,侵袭周围重要结构,包括蝶鞍、海绵窦、颅内、斜坡、鼻旁窦等,IPA约占肿瘤的25%~55%[3-5]。1993年,Knosp提出基于MRI冠状位颈内动脉与肿瘤的关系来判断肿瘤是否侵袭海绵窦,被广泛用于IPA的影像学评估。与Knosp分级相比,Hardy分级系统中肿瘤向鞍上部分扩展是否代表肿瘤侵袭一直有争议。同时,肿瘤侵袭机制的研究也发现,肿瘤侵袭与基质金属蛋白酶(matrix metalloproteinases,MMPs)、垂体肿瘤转化基因(pituitary tumor transforming gene,PTTG)、血管内皮生长因子(vascular endothelial growth factor,VEGF)等分子相关,可以作为肿瘤侵袭的分子标志物。但进一步研究发现,这些标志物在临床应用中并不能可靠地预测肿瘤预后,很多侵袭性腺瘤预后良好。2004年版WHO垂体肿瘤分类将IPA定义为放射学、术中肉眼观察和组织学观察到肿瘤对颅骨、神经和血管的侵犯,但显微镜下常见的硬脑膜侵犯并不能作为肿瘤侵袭性生长的指标。目前,Knosp分级和术中或术后病理切片观察到肿瘤对周围结构的侵袭仍是诊断IPA的基础。2013年,Trouillas等[6]采用了一种新的垂体腺瘤分类,根据肿瘤侵袭和增殖的特性将腺瘤分为5级(表4),依据此分级对410例患者进行8年的临床观察,发现2b分级以上的肿瘤预后较差。新版WHO分类仍然未把IPA作为病理诊断纳入分级,Lopes[7]指出,在WHO会议上很多研究者力主将IPA纳入WHO分类,但最终未能达成共识,主要原因是IPA的诊断依赖放射学、术中所见和病理学表现的综合判断,诊断标准不够客观、精确,病理医生难以单独做出诊断。但在新版分类中仍认为侵袭行为是预测肿瘤预后的重要因素之一。
鉴于IPA的生物学行为介于非侵袭性垂体腺瘤和垂体癌之间,2004年版WHO垂体肿瘤分类提出“非典型垂体腺瘤”的概念,将垂体腺瘤分为3类,包括垂体腺瘤、非典型垂体腺瘤和垂体癌,其中非典型腺瘤的诊断标准包括核分裂像增多、Ki-67增殖指数>3%和p53核染色阳性,试图从组织病理学表现预测垂体腺瘤的生物学行为。按照此标准,非典型腺瘤的发生率约为5%~10%,但经过10余年的临床观察,很多研究发现该分类并不能很好地预测患者的预后。同时,很多研究者对于Ki-67指数的计数标准和p53染色的作用也提出质疑,如2015年Chiloiro等[8]和2016年Miermeister等[9]发表的两篇关于非典型垂体腺瘤的研究,前者Ki-67指数的标准采用的是1.5%,后者采用的是4%,两篇文章均认为自己的标准可以区分典型和非典型垂体腺瘤。p53染色阳性的界定更是有赖于病理医生的主观判断,不同研究间差异很大,很多学者质疑p53阳性对垂体腺瘤预后的判断作用。因此,新版WHO垂体腺瘤分类取消了非典型垂体腺瘤这一分类,但保留了Ki-67指数和p53作为评估肿瘤增殖的分子标志物。
如前文所述,IPA不适合纳入肿瘤分类,非典型垂体腺瘤分类亦不够准确,对于巨大侵袭、易复发、增殖快、预后差的垂体腺瘤分类成为人们关注的问题,很多学者重新定义了CAPA的概念[10-13]。与IPA的认定主要依赖影像学、术中所见和组织病理学表现不同,CAPA的定义主要依据肿瘤对临床治疗手段的反应,包括对手术、放疗和化疗的抵抗,即传统治疗手段疗效较差,难以治愈,因此笔者将其翻译为难治性垂体腺瘤。该类肿瘤约占垂体腺瘤的10%,目前仍无统一标准将其准确定义,在一些文献中有学者将其与IPA混用。新版WHO分类并未将其引入分级系统来替代非典型垂体腺瘤,甚至未给出CAPA的准确定义,只是在描述中将肿瘤增殖(有丝分裂计数和Ki-67指数)和侵袭作为CAPA的重要标志物,其他指标包括促肾上腺皮质激素(ACTH)细胞腺瘤和PIT1阳性的多激素腺瘤中MGMT启动子甲基化和表达缺失。同时指出疏颗粒型GH细胞腺瘤、男性催乳素(PRL)细胞腺瘤、嗜酸干细胞腺瘤、Crooke细胞腺瘤、静默型ACTH细胞腺瘤中CAPA的比例更高。新版分类中还提到高风险垂体腺瘤(high-risk pituitary adenoma),其特征包括快速生长、影像学可见侵袭、高Ki-67指数、易于复发、传统治疗效果较差,其实也是CAPA的范畴。既往研究中提出的CAPA的分子标志物[10]多数存在争议或未经过大宗病例验证,未被新版分类系统采用。
总之,IPA、非典型垂体腺瘤和CAPA都是为了描述表现为部分恶性肿瘤行为、预后较差的垂体腺瘤。2004年版WHO分类中提出的非典型垂体腺瘤是一次不成功的尝试,新版WHO分类仍未给出这部分肿瘤的准确定义,有待于进一步的研究阐述肿瘤的进展机制,并发现新的分子标志物。
表4. Trouillas 垂体腺瘤的临床病理分类[6]
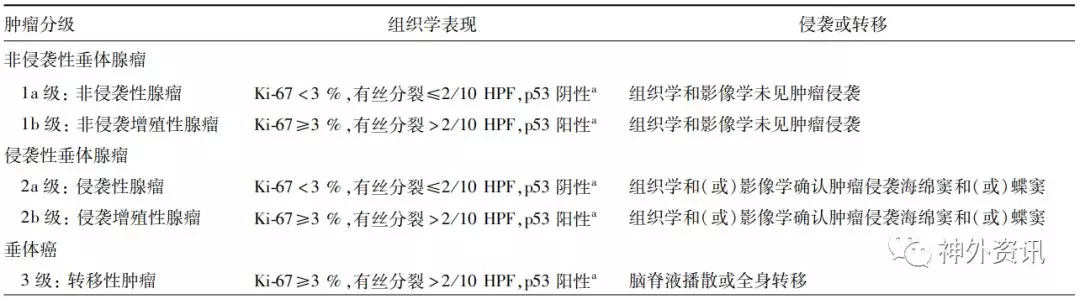
注:a甲醛固定样本Ki-67指数应≥3 %;Bouin-Hollande固定液固定样本Ki-67指数应>1 %;p53阳性标准为>10个核染色强阳性/10 HPF,增殖活性肿瘤必须符合3条标准中的至少2条;HPF:高倍镜视野(0.30 mm²、400×),修改自Trouillas[6]
二、垂体非神经内分泌肿瘤分类
新版分类对垂体非神经内分泌肿瘤也进行了详细地阐述(表5),较前版添加了很多种类,包括发生于鞍区的颅咽管瘤、神经元和副神经元肿瘤、垂体后叶肿瘤、间叶组织肿瘤、生殖细胞肿瘤、淋巴造血系统肿瘤和继发肿瘤等,但多数与2016版中枢神经系统肿瘤分类或其他系统肿瘤的WHO分类保持一致。变化较大的是新增了垂体母细胞瘤(pituitary blastoma)这一诊断,该肿瘤是发生于24个月以下(中位年龄为8个月)婴幼儿的一种罕见恶性肿瘤,由DICER1基因突变引起,常有库欣病的症状和体征。另外,对发生于垂体后叶的垂体细胞瘤、颗粒细胞瘤和梭形细胞嗜酸细胞瘤,新版认为这3种肿瘤均表达甲状腺转录因子1(thyroid transcription factor 1 ,TTF1),可能来源于垂体后叶的同一种细胞。同时,垂体后叶肿瘤还增加了鞍区室管膜瘤这一诊断。
三、小结与展望
新版WHO垂体肿瘤分类是对目前广泛应用的垂体肿瘤临床功能分型的进一步深化和扩充,遵循从肿瘤细胞起源角度命名的原则,将原有垂体腺瘤分类进行了重新阐述,同时调整或增加了一些新的垂体肿瘤分类,反映了近年来临床医生、病理医生和基础研究相关专家对垂体肿瘤研究的进展和思考,具有很强的临床可操作性和很高的临床实用价值。但也应认识到,当前许多肿瘤的分子病理分类已经非常成熟,在肿瘤分子水平揭示肿瘤发生机制和预测预后是大多数肿瘤研究的方向。但对于垂体腺瘤来说,除了一些综合征有关的肿瘤致病基因已被确定外,目前只发现GNAS基因和USP8基因突变与散发肿瘤的发生有关。因此,垂体肿瘤分子分类的研究仍任重道远,继续加强垂体肿瘤的分子病理学研究,在分子水平阐释肿瘤的发生机制并进行分子分类,是垂体肿瘤分类的最终目标,此次垂体肿瘤分类的更新仅仅是一个开始。
表5. 2017版与2004版垂体非神经内分泌肿瘤分类比较

注:a具体分类同2016版《WHO中枢神经系统肿瘤分类》;b包括软骨瘤、纤维瘤、血管球瘤、血管母细胞瘤、脂肪瘤、黏液瘤、软骨肉瘤、纤维肉瘤、平滑肌肉瘤、骨肉瘤和横纹肌肉瘤
参考文献
[1]Lloyd RV,Osamura RY,Kloppel G,et al.WHO classification of tumours of endocrine organs[M]. 4th edn,Lyon: International Agency for Research on Cancer(IARC) Press,2017:11-64.
[2]Nishioka H, Inoshita N, Mete O,et al. The Complementary Role of Transcription Factors in the Accurate Diagnosis of Clinically Nonfunctioning Pituitary Adenomas[J]. Endocr Pathol,2015,26(4):349-355. DOI: 10.1007/s12022-015-9398-z.
[3]Scheithauer BW, Kovacs KT, Laws ER, et al. Pathology of invasive pituitary tumors with special reference to functional classification[J]. J Neurosurg, 1986,65(6):733-744. DOI: 10.3171/jns.1986.65.6.0733.
[4]Thapar K, Kovacs K, Scheithauer BW, et al. Proliferative activity and invasiveness among pituitary adenomas and carcinomas: an analysis using the MIB-1 antibody[J]. Neurosurgery, 1996,38(1):99-106; discussion 106-107.
[5]Meij BP, Lopes MB, Ellegala DB, et al. The long-term significance of microscopic dural invasion in 354 patients with pituitary adenomas treated with transsphenoidal surgery[J]. J Neurosurg, 2002,96(2):195-208. DOI: 10.3171/jns.2002.96.2.0195.
[6]Trouillas J, Roy P, Sturm N, et al. A new prognostic clinicopathological classification of pituitary adenomas: a multicentric case-control study of 410 patients with 8 years post-operative follow-up[J]. Acta Neuropathol, 2013,126(1):123-135. DOI: 10.1007/s00401-013-1084-y.
[7]Lopes MBS. The 2017 World Health Organization classification of tumors of the pituitary gland: a summary[J]. Acta Neuropathol, 2017,134(4):521-535. DOI: 10.1007/s00401-017-1769-8.
[8]Chiloiro S, Doglietto F, Trapasso B, et al. Typical and atypical pituitary adenomas: a single-center analysis of outcome and prognosis[J]. Neuroendocrinology, 2015,101(2):143-150. DOI: 10.1159/000375448.
[9]Miermeister CP, Petersenn S, Buchfelder M, et al. Erratum: Histological criteria for atypical pituitary adenomas--data from the German pituitary adenoma registry suggests modifications[J]. Acta Neuropathol Commun, 2016,4:21. DOI: 10.1186/s40478-016-0290-y.
[10]Chatzellis E, Alexandraki KI, Androulakis II, et al. Aggressive pituitary tumors[J]. Neuroendocrinology, 2015,101(2):87-104. DOI: 10.1159/000371806.
[11]Di IA, Rotondo F, Syro LV, et al. Aggressive pituitary adenomas--diagnosis and emerging treatments[J]. Nat Rev Endocrinol, 2014,10(7):423-435. DOI: 10.1038/nrendo.2014.64.
[12]Raverot G, Castinetti F, Jouanneau E, et al. Pituitary carcinomas and aggressive pituitary tumours: merits and pitfalls of temozolomide treatment[J]. Clin Endocrinol (Oxf), 2012,76(6):769-775. DOI: 10.1111/j.1365-2265.2012.04381.x.
[13]Zaidi HA, Cote DJ, Dunn IF, et al. Predictors of aggressive clinical phenotype among immunohistochemically confirmed atypical adenomas[J]. J Clin Neurosci, 2016,34:246-251. DOI: 10.1016/j.jocn.2016.09.014.
