

提示
“浙二神外周刊”不定期接收外院投稿,审核后发表。欢迎各专业同道联系我们,分享精彩病例、研究热点或前沿资讯。投稿请联系:shishi74@163.com
前言
2018年5月27日由默沙东公司承办,在杭州召开的由华山医院、上海市一医院,苏州大学附属一院,福州军区总院,浙江省肿瘤医院及浙医二院参加的华东片区中国胶质瘤MDT团队交流会上,各团队提供了许多很好的病例,各有特色。专家讨论热烈,精彩纷呈。参会者普遍反映获益匪浅。现由《浙二神外周刊》及《神外资讯》分期刊出,与大家共享。
本病例由浙江大学医学院附属第二医院胶质瘤MDT团队( 神经外科: 徐锦芳 沈宏 放疗科:魏启春; 病理科:李百周;影像科:蒋飚)提供。
病史简介
患者,男性,36岁。因“头晕2年,加重3月余”入院。
患者2年前无明显原因出现头晕,无头痛,症状不重,间断性发作,休息能好转,无恶心呕吐,无肢体抽搐。未予特殊治疗。3月余前患者头晕症状加重,持续时间较长,当地医院头颅CT检查,提示左侧额颞区占位性病变,肿瘤可能(图1)。
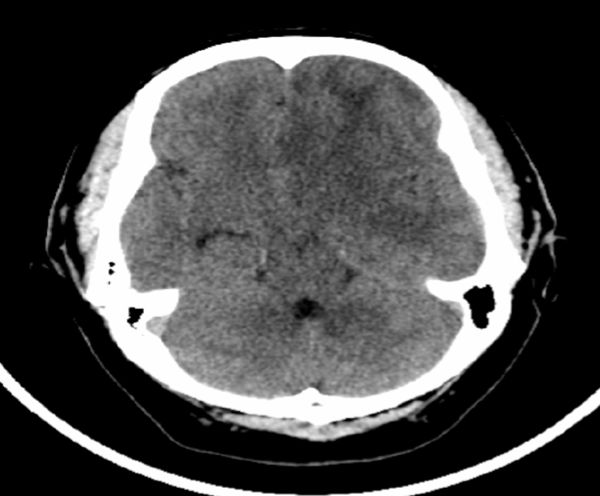
图1. 头部CT显示左额颞区低密度占位性病变,肿瘤可能
入院查体:神志清,对答如常,肢体活动自如,神经系统检查无明显阳性体征。
进一步MRI(包括DWI,MRS及DTI)检查,显示病灶弥漫性位于左额、颞及岛叶,T1低信号,T2高信号,无明显增强。考虑为低级别胶质瘤可能(图2)。
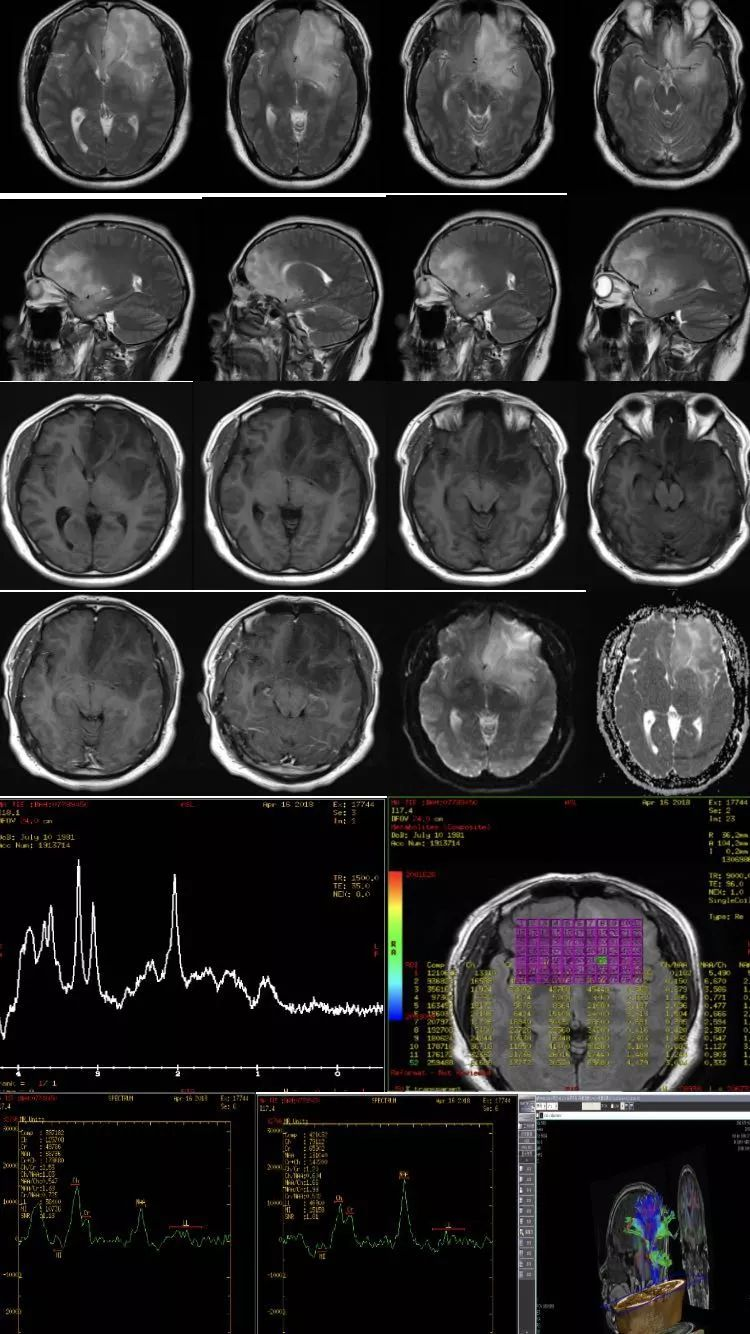
图2. MRI显示左额、颞及岛叶弥漫性胶质瘤
诊疗经过
入院后完善各项辅助检查等,应用神经电生理监测及术中磁共振辅助显微镜下开颅肿瘤切除术(图3)。术中见肿瘤质地软,血供一般,无明确边界。额叶、颞叶肿瘤基本全切除,岛叶肿瘤次全切除。手术经过顺利,术后恢复良好。
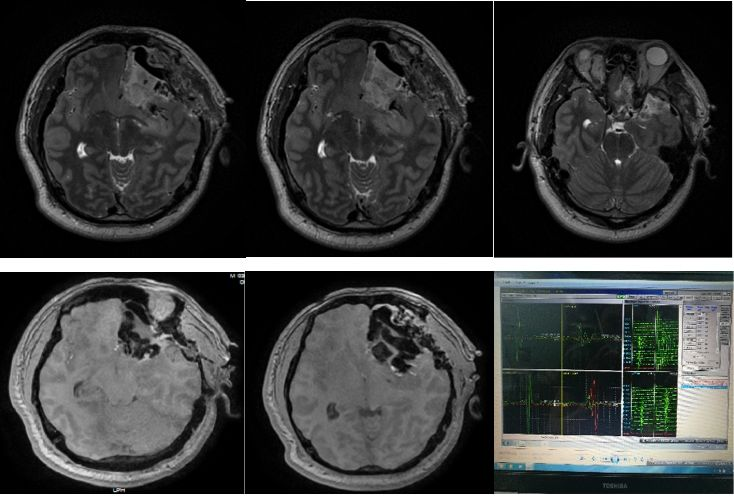
图3. 术中MRI检查显示岛叶后部残留,进一步再切除。神经电生理监测无异常改变
患者术后一般情况良好,无新增神经系统功能障碍。术后一天MRI复查,显示除岛叶后部少量残留外,余基本切除(图4)。
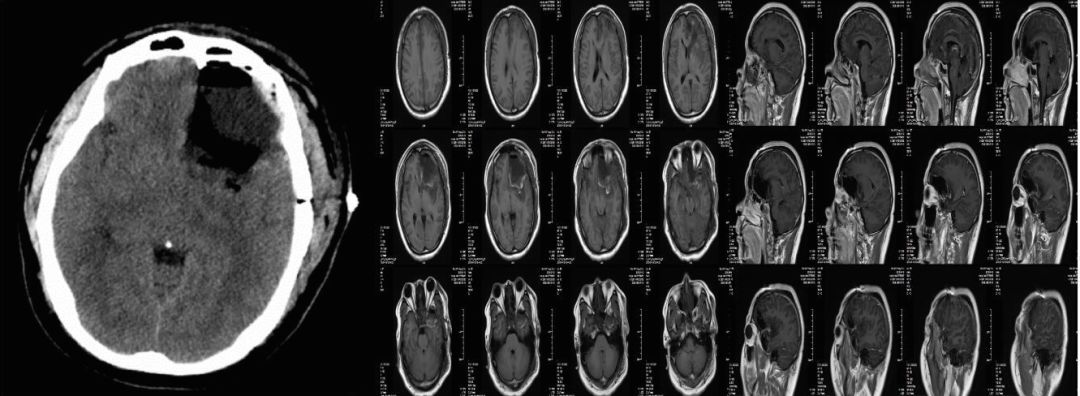
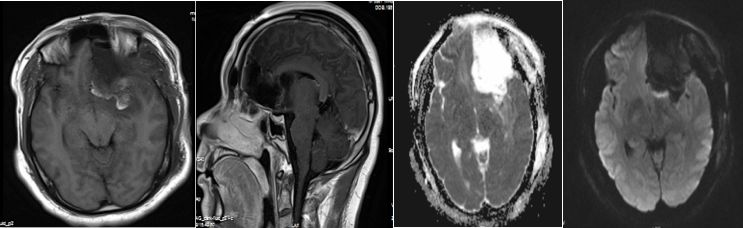
图4.术后一天CT/MRI复查,显示除岛叶后部少量残留外,余基本切除。
病理结果
术后病理回报:(左额颞岛叶)间变性少突胶质细胞瘤,WHO III级,IDH突变型;免疫组化结果:IDH1+,ATRX+,P53-,BRAF-V600E-,H3K24M-,Ki-67 10%;分子检测结果:(FISH)符合1p/19q杂合性共缺失(图5)。
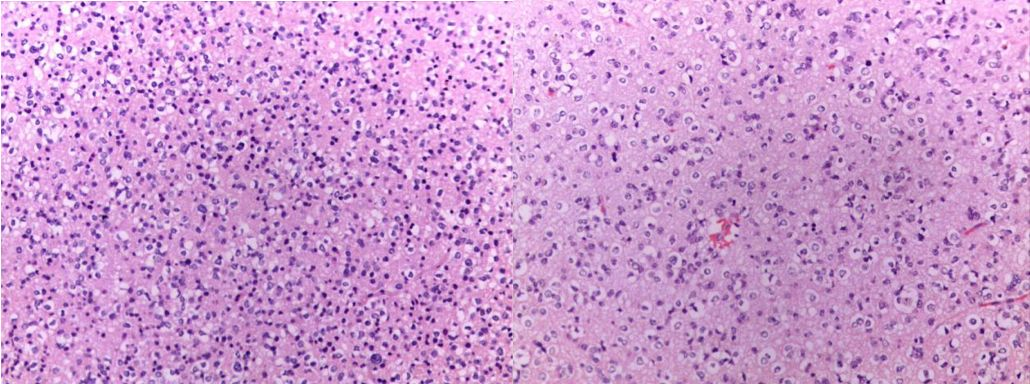
图5. 病理结果:左额,颞,岛叶间变少突胶质细胞瘤
讨论(专家点评诊断分析及诊疗意见)
1、关于此患者的病理诊断
此患者术后病理诊断明确,没有疑问。
根据2016 WHO中枢神经系统肿瘤分类,CNS肿瘤原先分7大类肿瘤,现被拆分为17大类肿瘤。原属于神经上皮起源肿瘤中的胶质瘤和胚胎性肿瘤变动最大[1]。
胶质瘤的分类、命名及诊断原则:(1)按肿瘤分子遗传学特征进行归类,如将IDH基因突变相关的星形细胞肿瘤和少突胶质细胞肿瘤归为一类,而把与IDH基因无关的毛细胞型星形细胞瘤等归入其他星形细胞肿瘤;(2)定义特定胶质瘤类型应包含分子遗传学特征,如少突胶质细胞瘤,IDH突变及1p/19q共缺失型;(3)命名法采用组织学分型+分子亚分型,如胶质母细胞瘤,IDH野生型;(4)在组织学分型和分子亚分型不一致时,强调分子亚分型胜过组织学分型,如组织学表现为星形细胞瘤,但有IDH突变及1p/19q共缺失,应"整合"诊断为"少突胶质细胞瘤,IDH突变及1p/19q共缺失型";(5)2016修订版并未摒弃原有的组织学分型,在不具备条件或未能获得足够分子检测信息时,仍采用原有的组织学分型,但须用“非特指”(NOS)说明此类病理诊断未能确定分子亚型;(6)为使病理诊断更规范和标准化,提出病理诊断"分层化"理念,包括:WHO组织学分类和分级及分子检测结果,综合三方面信息做出整合诊断;(7)2016修订版并未特别规定何种技术或方法用于肿瘤分子检测,因此,建议在病理报告中标明分子检测方法及结果。[2,3,4]
大脑胶质瘤病的分子遗传学变异特征与弥漫浸润性胶质瘤无明显差异,现被认为是一种特殊的生长模式,常在间变性星形细胞瘤中出现,且大多数病例为IDH野生型,因此删除了该肿瘤类型。[5,6]
2、关于此患者的影像学诊断
此患者术前MR T1增强扫描显示隐约有条索状强化,因此术前影像提示可能不是低级别胶质瘤。
如何在整合了分子基因型的弥漫型胶质瘤分类和分级诊断中继续发挥作用,并找到合适的影像学标记物对IDH基因突变亚型做出预测,是影像医学发展目前亟待解决的问题。
MRI研究提示IDH基因突变型胶质瘤较野生型具有一定的特征性,如胶质瘤氢质子MRS研究显示IDH突变的下游代谢产物2-羟基戊二酸(2-hydroxyglutarate,2-HG)在2.25ppm位置有特异性的波峰出现(图2),而且通过定量分析显示IDH2突变相比IDH1突变产生了更多的2-HG。另外,弥漫型胶质瘤MR灌注加权成像研究也表明较低的相对脑血容量(relativecerebralblood volume, rCBV)是IDH 突变型弥漫型胶质瘤(WHO Ⅱ、Ⅲ级)区别于野生型的重要功能影像特征,利用rCBV可以对弥漫型胶质瘤(WHOⅡ、Ⅲ级)的IDH突变亚型做出预测。
尽管大脑胶质瘤病作为一个独立的诊断病种已从2016 CNS WHO 肿瘤分类中删除,但其作为一种生长模式,却存在于多种胶质瘤中,包括IDH突变型星形细胞瘤和少突胶质细胞瘤以及IDH野生型胶质母细胞瘤。根据MR检查中肿瘤是否强化以及扩散受限程度可以对WHO Ⅱ级和Ⅲ级弥漫型星形细胞瘤进行准确分级。有研究显示MR灌注加权成像的rCBV 值预测IDH突变型胶质瘤的准确性为88%(WHO Ⅱ、Ⅲ级),其主要原理为IDH突变代谢物2-HG 与保持低氧诱导因子(hypoxia-inducingfactor-1A,HIF-1A)的低水平有关;而HIF-1A 是肿瘤血管生成的重要诱导因子,因此,IDH突变的胶质瘤可能具有比野生型相对较低的rCBV水平。更近的一项研究提示,扩散加权成像技术(如DTI和DWI)可以鉴别不同级别弥漫型星形细胞瘤的IDH1(R132H)突变状态。[7]
3、关于此患者的手术治疗
在不损伤功能的前提下最大程度地安全切除肿瘤是累及功能区低级别胶质瘤手术的关键。术前根据多模态影像学检查、神经功能、神经心理学等评估,综合考虑制定手术计划,选择术中功能监测。推荐术中唤醒下皮质及皮质下直接电刺激、神经导航、术中MRI、术中超声以及皮质体感诱发电位定位中央沟,运动诱发电位监测运动区等辅助技术。以避免术后永久性神经功能损伤的发生,显著提高患者术后的生存质量。[8]
4、关于此患者的后续放化疗治疗
脑胶质瘤的遗传学成分对于预测胶质瘤患者的预后具有重要的帮助。低级别胶质瘤的分子病理特点为:IDHl/2突变(50%~80%)、TP53突变(星形细胞瘤中50%~60%)、ATRX缺失(星形细胞瘤中约70%)以及1p/19q联合缺失(少突胶质细胞瘤中30%一60%)。这些分子标志物对患者的生存预后具有重要意义。
对于高级别胶质瘤尤其是胶质母细胞瘤,推荐术后应尽早 (<6周)进行放疗。并建议替莫唑胺化疗。还要注意此患者病理类型可能通过脑脊液播散和种植。[9]
5、关于此患者的随访
患者的术后评价过程推荐采用神经影像学与行为量表相结合的方式。建议患者在术后72 h内及3个月行平扫及增强磁共振T2/FLAIR检查,以评价肿瘤切除情况及作为影像学随访的参考标准。推荐在术后2周、3个月、6个月、12个月分别评价患者的KPS评分、语言功能、运动功能及生命质量等。强烈建议患者5年内每3~6个月复查磁共振成像,观察判断肿瘤是否复发。
参考文献
[1]LouisDN, OhgakiH, WiestlerOD, et al. World Health Organizationclassification of tumours of the central nervous system[M]. Lyon: IARC Press,2016.
[2]PerryA. WHO s arrived in 2016! An updated weather forecast for integratedbrain tumor diagnosis[J]. Brain Tumor Pathol, 2016, 33(3):157-160. DOI:10.1007/s10014-016-0266-4.
[3]LouisDN, PerryA, ReifenbergerG,et al. The 2016 World Health Organization Classificationof Tumors of the Central Nervous System: a summary[J]. Acta Neuropathol, 2016,131(6):803-820. DOI: 10.1007/s00401-016-1545-1.
[4]BroniscerA, ChamdineO, HwangS,et al. Gliomatosis cerebri in children shares molecularcharacteristics with other pediatric gliomas[J]. Acta Neuropathol,2016,131(2):299-307. DOI:10.1007/s00401-015-1532-y.
[5]HerrlingerU, JonesDT, GlasM, et al.Gliomatosis cerebri: no evidence fora separate brain tumor entity[J]. Acta Neuropathol, 2016,131(2):309-319. DOI:10.1007/s00401-015-1495-z.
[6]中华医学会病理学分会脑神经病理学组.2016 W HO 中枢神经系统肿瘤分类第4版修订版概述及胶质瘤部分介绍[J].中华病理学杂志,2016,(11):745-747.DOI:10.3760/cma.j.issn.0529-5807.2016.11.001.
[7]任彦,李安宁,吴越, 等.从影像医学角度解读2016 WHO中枢神经系统肿瘤分类[J].中华放射学杂志,2016,(11):811-816. DOI:10.3760/cma.j.issn.1005-1201.2016.11.002.
[8]《中国中枢神经系统胶质瘤诊断和治疗指南》编写组.中国中枢神经系统胶质瘤诊断与治疗指南(2015)[J].中华医学杂志,2016,(7):485-509.DOI:10.3760/cma.j.issn.0376-2491.2016.07.003.
[9]中华医学会放射肿瘤治疗学分会.胶质瘤放疗中国专家共识(2017)[J].中华放射肿瘤学杂志,2018,(2):123-131. DOI:10.3760/cma.j.issn.1004-4221.2018.02.001.
(本文由浙二神外周刊原创,浙医二院神经外科徐锦芳副主任医师整理并审校,张建民主任终审)

